




According with different international organisations, cardiovascular diseases are becoming the first cause of death in western countries. Although exposure to different risk factors, particularly those related to lifestyle, contribute to the etiopathogenesis of cardiac disorders, the increase in average lifespan and ageing are considered major determinants of cardiac diseases events. Mitochondria and oxidative stress have been pointed out as relevant factors both in heart ageing and in the development of cardiac diseases such as heart failure, cardiac hypertrophy and diabetic cardiomyopathy. During ageing, cellular processes related with mitochondrial function, such as bioenergetics, apoptosis and inflammation are altered leading to cardiac dysfunction. Increasing our knowledge about the mitochondrial mechanisms related with the ageing process, will provide new strategies in order to improve this process, particularly the cardiovascular ones.
De acuerdo con diferentes organizaciones como la Asociación Americana del Corazón o la Organización Mundial de la Salud, las enfermedades cardiovasculares se han convertido en la primera causa de muerte en países occidentales. Aunque la exposición a diferentes factores de riesgo, en particular los relacionados con el estilo de vida, contribuyen de manera significativa a la etiopatogénesis de enfermedades cardíacas, el incremento en la esperanza de vida y el envejecimiento de la población asociado a él se consideran los determinantes principales del inicio y desarrollo de las mismas. Las mitocondrias y el estrés oxidativo se han señalado como factores relevantes tanto en el envejecimiento del corazón como en el desarrollo de enfermedades cardíacas como la insuficiencia cardíaca, la hipertrofia cardíaca y la miocardiopatía diabética. Durante el envejecimiento, diferentes procesos celulares relacionados con la función mitocondrial, como la bioenergética, procesos de apoptosis o de inflamación, se ven alterados, lo que conlleva una reducción en la supervivencia celular, y como consecuencia, disfunción cardíaca. Aumentar nuestro conocimiento sobre los mecanismos mitocondriales relacionados con el proceso de envejecimiento proporcionará nuevas estrategias para mejorar de forma más eficiente este proceso y las diferentes enfermedades relacionadas con él, en particular las cardiovasculares.
According to the World Health Organization, cardiovascular diseases are now the leading cause of death in western countries,1 replacing neurodegenerative disorders. In Europe, despite the fact that the number of deaths related to cardiovascular diseases has decreased over recent years, these diseases are still responsible for the death of around 4million people, almost 50% of all deaths in Europe. If we look at current statistics, Spain is high on the list of European countries with lower rates of mortality due to coronary heart disease, the number one cause of death among all cardiovascular diseases.2
Although certain environmental and lifestyle-related factors, such as diet and physical inactivity, play a major role in the aetiopathogenesis of heart disorders, ageing is considered the main determinant of heart disease. However, other age-related diseases, such as dyslipidaemia and diabetes mellitus, exacerbate the negative effects of ageing on the cardiovascular system. According to the Instituto Nacional de Estadística (Spanish National Institute of Statistics), life expectancy in Spain has increased an average of one year every five years since 1975. Also, over the last 20 years, life expectancy at birth has increased by approximately 5 years. This increase has a direct consequence: increase in incidence rates of age-related diseases, especially cardiovascular diseases. More importantly still, this increase in average life expectancy is expected to continue to rise over the next 20 years, when around 20% of the population will be aged 65 or older. The heart is primarily a post-mitotic tissue and exhibits a highly aerobic metabolism. These features implicate high dependence on mitochondrial function for proper functioning of the heart cells.3 Mitochondria play a determining role in the function and survival of cardiomyocytes and are fundamental for meeting the high energy demands of the myocardium. Under physiological conditions, 20–30% of the total cardiomyocyte volume is occupied by mitochondria, but this value may increase when the energy requirements of the myocardium increase. The heart consumes the equivalent of 6kg of ATP per day, the majority of which is generated through mitochondrial oxidative phosphorylation that is fuelled by catabolism of lipids and carbohydrates.4 The heart's energy reserve includes ATP (≈5μmol/g wet weight) and phosphocreatine (PCr; ≈8μmol/g wet weight), with the latter acting as a transport system and ATP buffer.5 In mitochondria, the phosphate group of ATP can be transferred to creatine by mitochondrial creatine kinase to form PCr. PCr can easily diffuse through the mitochondrial membrane into the cytosol, where it can be used to generate ATP from ADP through reactions catalysed by the cytosolic creatine kinase.6 There is a delicate balance between nuclear and mitochondrial gene expression, which regulates the assembly of mitochondrial respiratory complexes. Especially under exercise conditions, mitochondrial biogenesis is activated through modulation of the ATP:ADP ratio, activation of AMP-activated protein kinase, and the subsequent expression of factors PGC-1α and NRF-1.7 In turn, when energy demands increase, gene expression of nuclear and mitochondrial DNA (mtDNA) is activated, maximising the capacity of mitochondria to perform oxidative phosphorylation.
Oxidative stress and mitochondrial genomic instabilityConsidering that ageing is one of the main factors leading to impaired heart function, it is especially important to understand the mechanisms underlying the ageing process in order to find therapeutic approaches to improve this process. Various hallmarks of the ageing process have been considered, including mitochondrial dysfunction, genomic instability and epigenetic alterations.8 Since it was suggested as one of the main determinants of longevity,9 a large number of studies have supported the role of mitochondria and oxidative stress in the ageing process. Although free radicals are generated in different cell compartments and by multiple enzymes, such as NADPH oxidase or xanthine oxidase, mitochondrial reactive oxygen species (ROS) production is considered the most important source of free radicals in healthy tissues under physiological conditions. This is because the main free radical generator, the electron transport chain, is located in the inner mitochondrial membrane.10 These radicals are generated continuously during oxidative phosphorylation and ATP generation. For many decades, ROS have been primarily investigated for their role as oxidising molecules; however, these ROS also play a major role as signalling molecules and are involved in various physiological responses that control cellular homeostasis.11
Under physiological conditions, a variable amount of oxygen is converted to superoxide anion (O2−) due to electron leak predominantly from mitochondrial complexes I and III.10,12 Superoxide dismutase catalyses the reaction that transforms O2− into H2O2, which is then neutralised, resulting in O2 and H2O due to the activity of enzymes such as catalase. H2O2 can further be converted into hydroxyl radical (OH−) by Fenton and Haber–Weiss reactions in the presence of iron. Mitochondrial iron content increases significantly with ageing in various tissues, including the myocardium, which may be associated with an increase in OH− generation and therefore oxidative damage in old age.13,14 Continuous production of O2− and other ROS in the mitochondria leads to increased oxidative damage to various macromolecules with age. One type of damage produced by ROS that is especially relevant is damage to DNA. Due to this continuous exposure to free radicals and to different exogenous factors, the integrity of both nuclear and mitochondrial DNA is compromised during ageing.15,16 More specifically, mtDNA instability has been considered especially relevant in the ageing process since it leads to mitochondrial dysfunction.17–19 Free radicals generate a large number of mtDNA lesions, including oxidised DNA bases and simple- or double-strand breaks, the latter being some of the most harmful DNA lesions. Many of these ROS-induced DNA lesions show mutagenic or cytotoxic effects due to mispairing of bases, which may give rise to mutations upon DNA replication.20,21 Continuous DNA damage may cause blockage of replication and transcription processes, contributing to genomic instability. In order to prevent such instability, there are various DNA repair pathways in both the nucleus and the mitochondria. A particular repair pathway is activated depending on the type of DNA damage caused.22 However, despite the existence of these DNA repair pathways, DNA damage increases and accumulates with age. Therefore, the frequency of mtDNA point mutations and deletions is approximately 3-fold higher in the heart of aged mice than in young animals.23 The importance of cumulative mtDNA mutations as a determining factor in ageing and loss of function in various tissues, including heart dysfunction, has been confirmed in animal models with increased mtDNA instability. In one of these mouse models, mitochondrial transcription factor A was disrupted, specifically in the heart and muscle, resulting in a phenotype with respiratory chain deficiency and accumulation of morphologically abnormal mitochondria.24 These alterations were also associated with higher rates of apoptosis.25 Interestingly, these animals had normal heart function at birth but quickly developed dilated cardiomyopathy during the post-natal period.24 Another model is the PolG mouse, developed a decade ago.26,27 These mice have a high rate of mtDNA point mutations and deletions in all tissues,28,29 and are characterised by an early ageing phenotype. Cardiac mitochondria from these mice also show high levels of protein carbonyls and abnormal electron transport chain (ETC) activity, with depressed activity in the different ETC complexes.26 It has also been proposed that high levels of point mutations in mtDNA may affect the proper assembly of mitochondrial supercomplexes, leading to reduced ATP production and bioenergetic impairment.29 Moreover, PolG mice show increased apoptosis in cardiac tissue and premature age-related changes, such as cardiac hypertrophy and reduced systolic and diastolic function.23,30 Similarly, when these mice were subjected to endurance exercise, age-related changes, both in the skeletal muscle and heart, were delayed.31 The Safdar et al. study indicates that the effect of physical exercise may be at least partly mediated by induction of mitochondrial biogenesis, prevention of mtDNA mutations and reduced apoptosis.31 Recently, a new animal model has been developed showing increased accumulation of mtDNA deletions in the myocardium.32 Like PolG animals, these mice accumulate cardiomyocytes with compromised mitochondrial function, which may be related to the premature cardiac arrhythmia they develop.
One of the main consequences of mtDNA instability is mitochondrial dysfunction, which, as mentioned above, is thought to significantly contribute to the ageing process. However, mitochondria are not only critical for generating cellular energy via oxidative phosphorylation, but also participate in an important number of cellular functions, including apoptosis, Ca2+ homeostasis and redox signalling. Consequently, impaired mitochondrial function leads to loss of cellular homeostasis and therefore to tissue dysfunction.33,34 In addition to the accumulation of mtDNA mutations, other mechanisms also contribute to the age-related loss of mitochondrial function, such as oxidation of mitochondrial proteins, alterations in the lipid composition of mitochondrial membranes or alterations in mitophagy processes.8 It must be mentioned that mitochondrial function is not only affected by loss of mtDNA integrity. Since most of the mitochondrial proteins are encoded by nuclear DNA, increased nuclear genomic instability also contributes to impairment of mitochondrial function.35
Metabolic changes in the heart with ageingThe main metabolic pathway used by the heart is long-chain fatty acid β-oxidation which generates 70–90% of cardiac ATP.36 The remaining ATP comes from the oxidation of glucose and lactate, as well as small amounts of ketone bodies and certain amino acids.37 Use of fatty acids or glucose as substrate may directly inhibit the use of the other substrates (the Randle cycle). Substrate selection and interaction between glucose and fatty acids used in the heart have been considered highly relevant in the development of heart diseases since obtaining ATP from glucose is more efficient than from fatty acids.38 When it comes to mitochondria, levels of respiratory proteins and other key proteins involved in mitochondrial metabolism, including those associated with fatty acid metabolism, decline in the heart with ageing. On the contrary, proteins involved in glucose metabolism and certain extracellular structural proteins increase significantly with age.39 Therefore, increased expression of glycolytic proteins, together with a decline in fatty acid oxidation, would indicate metabolic remodelling with age similar to heart failure (HF) in younger individuals.40 Various alterations related to mitochondrial metabolism have been associated with the normal ageing process of the heart.41,42 For example, it has been proposed that HF is associated with a reactive hyperadrenergic state that increases circulating plasma free fatty acids, which leads to impaired glucose metabolism and insulin resistance.38 Likewise, age-associated pathophysiological changes in metabolism, such as diabetes, crucially contribute to the accumulation of oxidised proteins and advanced glycation/glycosylation end products in the heart.43 Some of the age-dependent cardiac alterations related to metabolic changes will be looked at later on in this review.
Cardiac remodelling during ageingMyocardial remodelling during ageing is related to changes in the amount and organisation of extracellular matrix components.44 Proper collagen arrangement prevents excessive fibre stretch, preserving heart function.45 One of the most recurrent and well-characterised alterations associated with cardiac ageing is the increased deposition of collagen within cardiomyocytes and around the blood vessels.46,47 During ageing, the rate of ventricular collagen turnover and synthesis by fibroblasts increases.48 Age-related fibrosis is characterised by increased collagen content, decreased collagen solubility and increased collagen fibre cross-linking.49 In addition to collagen accumulation, modifications and cross-linking of both collagen fibres and elastin contribute to the development of vascular calcification, which increases with ageing.50 Another extracellular matrix consequence is valve sclerosis, which affects more than 30% of elderly individuals. In fact, excessive increase of collagen fibres is associated with stiff ventricles and diastolic dysfunction in the aged heart. Likewise, in extracellular matrix destabilisation and cardiac remodelling, the relationship between matrix metalloproteinases and their endogenous tissue inhibitors plays a central role in stimulating collagen production. A recent study has shown that metalloproteinase-mediated degradation of elastin contributes to valve mineralisation and calcification by stimulating calcium deposition onto fragmented elastin fibres in humans.51 One of the most common consequences of cardiac remodelling in ageing is left ventricular hypertrophy (LVH) characterised by thickening of the ventricular walls. Clinical trials such as ‘The Framingham Heart Study’ or ‘The Baltimore Longitudinal Study on Aging’ have demonstrated an age-dependent increase in LVH in healthy adults without hypertension, a major risk factor for the development of LVH.52 With age, various mechanisms that promote cardiac hypertrophy are activated, such as mechanical stress, loss of cardiomyocytes and the previously described increased interstitial fibrosis.
One of the processes that promotes loss of cardiomyocytes is increased apoptosis during ageing. Inhibition of autophagy processes, partly due to accumulation of lipofuscin on lysosomes, is considered one of the main causes of increased apoptosis.53 Loss of autophagic capacity stimulates the accumulation of damaged and non-functional cell components, including organelles such as mitochondria, which activates apoptotic pathways.54 On the other hand, changes in myocytes may also cause alterations in Ca2+ handling. Intracellular Ca2+ plays a critical role in modulating cardiac function. Ca2+-induced Ca2+ release regulates myocardial contractility through activation of ion channels, activation of the ryanodine receptor and sodium-calcium exchanger activity. Sequestration of Ca2+ in the sarcoplasmic reticulum by the activity of the sarcoplasmic/endoplasmic reticulum calcium-ATPase (SERCA2a) pump triggers myocardial relaxation. In recent studies, it has been shown that the increased oxidative stress observed in the ageing heart also affects SERCA2a, decreasing its activity on Ca2+ and prolonging diastolic relaxation.55 Moreover, in a study conducted in female Fischer rats, a commonly used animal model of ageing-associated HF,56 prolonged cellular contractions in aged animals were observed to be caused by reduced transport of intracellular Ca2+ and increased expression of β-isoforms of myosin heavy-chain molecules.57,58 These modifications adversely affect electrical activity and the contractile power of cardiomyocytes. Boluyt et al. showed an alteration in the coupling-relaxation process of Ca2+-dependent myofilaments. The authors also described decreased phosphorylation of troponin i in myofilaments.56
Cardiac alterations during ageing. Role of mitochondriaDuring ageing, physiological processes decline progressively, decreasing homeostasis control and increasing morbidity. At the same time, the incidence of age-related diseases increases. Although all tissues are affected, those containing post-mitotic cells, such as the brain and heart, are considered to be especially affected.59 The cardiac ageing process implies various changes in the physiology and biochemistry of the heart and associated vessels. Morphologically, the heart undergoes thickening and hypertrophy of the left ventricle and interventricular septum. Stiffening, scarring, calcification of the aortic valve leaflets and aortic sclerosis increase. Electrical activity on the myocardium is also affected in the aged heart by calcification of the mitral valve and reduction of the number of pacemaker cells, due to apoptosis, at the sinoatrial and atrioventricular nodes along with accumulation of collagen, adipose tissue and amyloidosis.60,61
Cardiac hypertrophyCardiomyocytes can be damaged by different processes, such as ischaemia, presence of toxic substances and microorganisms. These pathological situations lead to inadequate contraction of the heart or loss of cardiomyocytes, with a compensatory response that is manifested by cardiac remodelling in the form of cardiac hypertrophy.62 Cardiac hypertrophy has traditionally been considered an adaptive response. During cardiac hypertrophy, protein synthesis intensifies, new sarcomeres are produced, cardiomyocytes become thicker and longer, the ventricular wall thickens and cardiac contractility increases. This response temporarily eliminates, or at least reduces, the haemodynamic overload of the heart.63,64 However, numerous studies have demonstrated that cardiac hypertrophy is associated with a significantly increased risk of diastolic dysfunction, HF and malignant arrhythmias.65,66 Pathological cardiac hypertrophy has been described as being associated with depletion of energy reserves, manifested as constant ATP levels and a reduction of PCr.67,68 The PCr/ATP ratio decreases and ATP decreases significantly as compensated hypertrophy advances to HF.69 In cultured cardiomyocytes, hypertrophy induced by angiotensin ii, endothelin 1, norepinephrine, TNF-α or mechanical stress has been associated with increased levels of oxidative stress and ROS-mediated activation of several intracellular signalling pathways, including NF-kB and mitogen-activated protein kinases.70 However, LVH caused by pressure overload is reduced when animals are treated with antioxidants.71
Heart failureAs mentioned above, although cardiac hypertrophy has traditionally been considered an adaptive response, several studies have demonstrated that it is associated with an increased risk of developing HF and malignant arrhythmias.65,72,73 HF is a growing public health problem, mainly due to the increased life expectancy of the population and its increased prevalence with age. In developing countries, around 2% of adults suffer from HF; the prevalence has been observed to increase to around 6–10% in individuals over the age of 65.74 The mechanisms involved in HF are complex and multifactorial. HF is characterised by a reduction in the rate or speed of myocardial relaxation, as well as decreased myocardial compliance.4,75 It is also associated with mitochondrial dysfunction and increased oxidative stress.76 Oxidative stress and alterations in intracellular Ca2+ homeostasis regulating proteins contribute to HF-related cardiac dysfunction.66 In addition, alterations in Ca2+ handling and myofilament function contribute to cardiac alterations. It has been suggested that mitochondria in endothelial cells may play an important role in cellular signalling as sensors for local oxygen concentration and regulators of nitric oxide production.77 Likewise, increased oxidative stress directly affects cardiomyocyte structure and function as it activates various signalling pathways involved in remodelling and HF.78 All of this indicates a close relationship between ROS production, mitochondrial dysfunction and the development of HF. Unlike diastolic dysfunction at rest, systolic function, as measured by ejection fraction, is preserved with age. HF with preserved ejection fraction (HFpEF) displays no cardiac dilation, although it is characterised by increased ventricular filling pressure, lung congestion, dyspnoea and exercise intolerance.79 The incidence of HFpEF has been seen to be increasing over recent years,80 particularly in elderly women, where 90% of new cases of HF are HFpEF.81 Although HFpEF was originally considered to be predominantly caused by diastolic dysfunction, more recent studies indicate that HFpEF in the elderly is characterised by a wide range of cardiac and non-cardiac abnormalities. In comparison with HF with reduced ejection fraction, the overall prognosis of HFpEF patients is similar to those with HF with reduced ejection fraction, although with a higher number of hospitalisations in patients in the first group. The need for effective therapeutic strategies for HFpEF has encouraged the development of suitable animal models for studying HFpEF. In these studies, aortic banding and systemic hypertension models have been widely used since hypertension contributes significantly to HFpEF.79 Another example is the Dahl salt-sensitive rat, which is characterised by hypersensitivity to sodium intake and is probably the most frequently used HFpEF animal model. When these animals are fed a high-salt diet (8% NaCl), they develop renal failure, hypertension (>175mmHg) and LVH, with HFpEF developing at just a few weeks of age.82 Moderate transverse-aortic constriction in young animals also triggers compensated concentric left ventricular hypertrophy, with particularly marked diastolic filling abnormalities. These abnormalities become progressively more exaggerated, representing a good model for studying HFpEF.
Diabetic cardiomyopathyIn obese people and those individuals with diabetes mellitus, cardiac dysfunction is considered a consequence of diabetic cardiomyopathy.83 Diabetic cardiomyopathy affects the myocardium of patients with diabetes and causes a broad spectrum of structural abnormalities resulting in LVH and systolic and diastolic dysfunction, or a combination of the two.84 The incidence of diabetes increases with age, and is especially relevant in the case of type 2 diabetes. The global prevalence of diabetes among adults aged 60 and older is 19% (approximately 135 million people) and accounts for 35% of all cases of diabetes in adults.85 Likewise, the incidence of coronary heart disease, stroke, congestive heart failure, hypertension, neuropathy, visual impairment and arthritis is higher among elderly adults with diabetes than in similar age adults without diabetes.86 We can differentiate between systemic and cardiac insulin resistance. Cardiac insulin resistance is defined as decreased insulin sensitivity or insulin-stimulated glucose uptake in the heart in the absence of systemic insulin resistance or risk factors for coronary heart disease such as obesity, hyperglycaemia, hyperinsulinaemia, hypercholesterolaemia and hypertension.87 Insulin resistance alone has severe adverse effects on cardiac function. In fact, the onset of hyperglycaemia and diabetes is often preceded by several years of insulin resistance. Studies carried out in mice with cardiomyocyte-specific knock-out of the insulin receptor (CIRKO) or insulin receptor substrate (CIRSKO) have investigated the relationship between insulin resistance and cardiac dysfunction. CIRKO and CIRSKO mice showed reduced insulin-stimulated glucose uptake and also impaired cardiac function. In glucose transporter 4 (GLUT4) knock-out mice, insulin resistance-associated cardiac dysfunction has also been observed.88–90 Insulin resistance in the presence of diabetes is associated with HF. Approximately 24% of all patients with HF and 40% of hospitalised patients with HF have diabetes mellitus and, over the next few decades, this percentage is expected to grow exponentially with the ageing of the population.91 Insulin resistance not only has negative effects on the heart, but also increases the risk of type 2 diabetes in both elderly men and women, which predisposes them to the development of coronary heart disease.92,93 Different mechanisms responsible for insulin resistance-induced cardiac dysfunction have been proposed. One such mechanism is endothelial dysfunction in response to insulin and decreased leucocyte telomere length.94,95 Given that mitochondria are the main source of ATP for meeting the energy demands of the heart, mitochondrial dysfunction has been considered as one of the underlying causes of metabolic disorder- and insulin resistance-related heart disease.96,97 Under normal physiological conditions, the heart uses energy from substrates (mainly fatty acids and carbohydrates) based on metabolic demand and availability.98 However, in the context of insulin resistance, the myocardium's ability to use glucose as an energy source is reduced.38,99 The pathophysiology of diabetic cardiomyopathy is closely associated with the change in substrate use.100 Diabetic cardiomyopathy has been shown to produce intracellular accumulation of toxic metabolic intermediates, such as long-chain acyl-CoA and acylcarnitine, which affect the mitochondrial ATP/ADP ratio, decreasing mitochondrial metabolic function.101
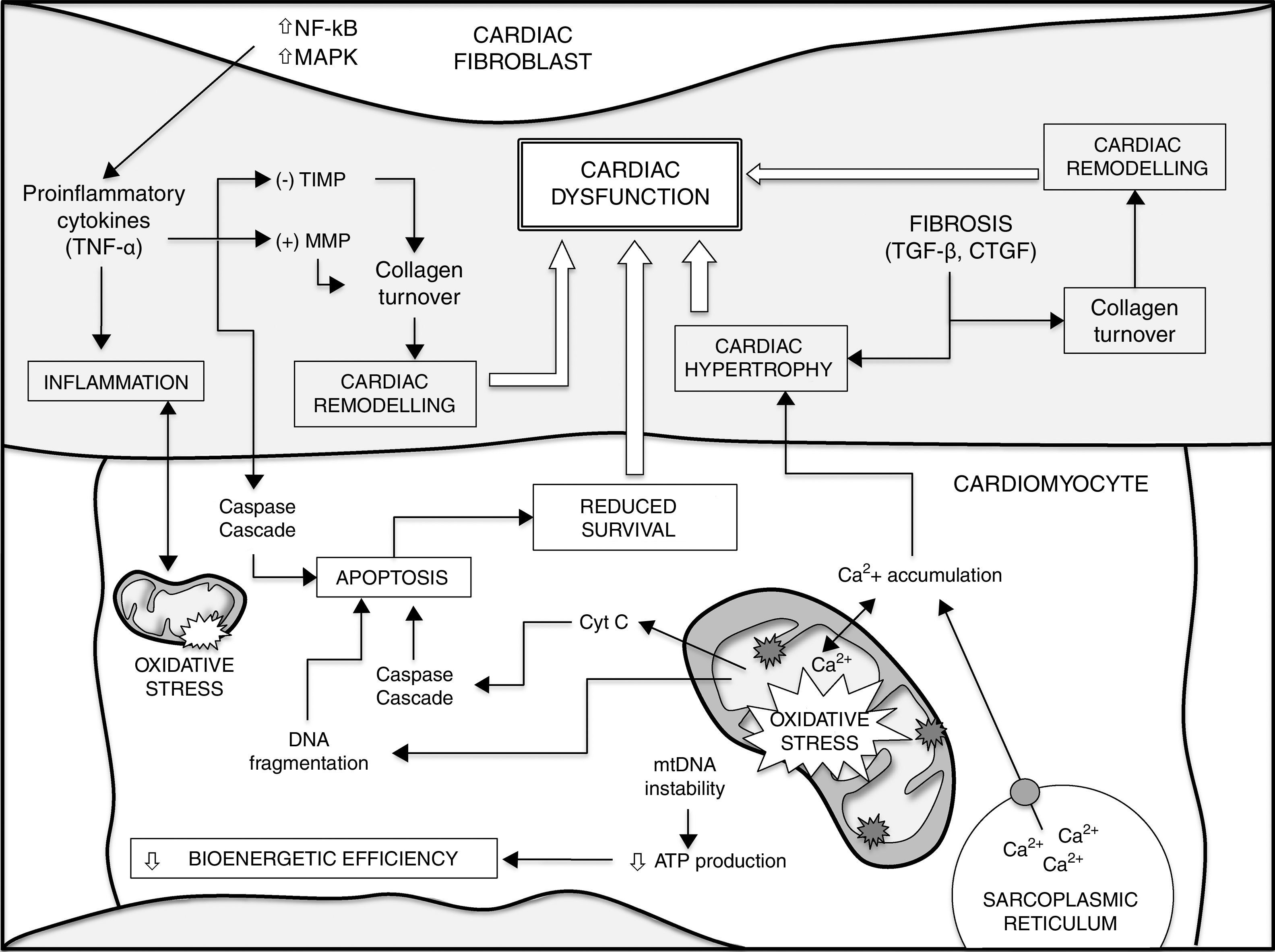
ConclusionsThe development of numerous heart diseases is associated with major changes in metabolic processes. Together with these changes, alterations in mitochondrial biogenesis and mitochondrial content in cardiomyocytes and the loss of cardiomyocyte function contribute to producing and aggravating a heterogeneous group of heart diseases. Mitochondrial ROS, accumulation of mtDNA mutations and loss of mitochondrial function play a fundamental role in loss of cardiomyocyte function and, therefore, cardiac ageing. Gaining a better understanding of fundamental mechanisms that determine the ageing process could lead to advances in the search for preventive and therapeutic treatments for heart disease. Fig. 1 shows a summary of the different factors that contribute to ageing of the heart and a higher susceptibility to developing heart disease.
. Increased mtDNA instability, enhanced apoptosis and loss of homeostasis (e.g. intracellular Ca2+ handling) in cardiomyocytes lead to impaired bioenergetic efficiency and reduced cell survival. On the other hand, the release of cytokines from cardiac fibroblasts and different growth factors, such as TGF-β, play a critical role in decreased cardiomyocyte survival during ageing, cardiac remodelling and fibrosis, contributing to cardiac hypertrophy. There is also a major positive correlation between oxidative stress and inflammatory processes. Finally, all these events lead to cardiac dysfunction. CTGF: connective tissue growth factor; MAPK: mitogen-activated protein kinase; MMP: matrix metalloproteinases; NF-κB: nuclear factor kappa-light-chain enhancer of activated B cells; TGF-β: transforming growth factor-beta; TIMP: endogenous tissue inhibitors of MMP; TNF-α: tumour necrosis factor alpha.")
Different factors contribute to ageing of the heart and a higher susceptibility to heart disease (see text for details and references). Increased mtDNA instability, enhanced apoptosis and loss of homeostasis (e.g. intracellular Ca2+ handling) in cardiomyocytes lead to impaired bioenergetic efficiency and reduced cell survival. On the other hand, the release of cytokines from cardiac fibroblasts and different growth factors, such as TGF-β, play a critical role in decreased cardiomyocyte survival during ageing, cardiac remodelling and fibrosis, contributing to cardiac hypertrophy. There is also a major positive correlation between oxidative stress and inflammatory processes. Finally, all these events lead to cardiac dysfunction. CTGF: connective tissue growth factor; MAPK: mitogen-activated protein kinase; MMP: matrix metalloproteinases; NF-κB: nuclear factor kappa-light-chain enhancer of activated B cells; TGF-β: transforming growth factor-beta; TIMP: endogenous tissue inhibitors of MMP; TNF-α: tumour necrosis factor alpha.
Both authors have contributed equally to this article.
Conflicts of interestThe authors declare that they have no conflicts of interest.
Please cite this article as: Martín-Fernández B, Gredilla R. Estrés oxidativo mitocondrial y envejecimiento cardíaco. Clin Investig Arterioscler. 2018;30:74–83.