





The very low-density lipoprotein receptor (VLDLR) plays an important function in the control of serum triglycerides and in the development of non-alcoholic fatty liver disease (NAFLD). In this study, we investigated the role of peroxisome proliferator-activated receptor (PPAR)β/δ activation in hepatic VLDLR regulation. Treatment of mice fed a high-fat diet with the PPARβ/δ agonist GW501516 increased the hepatic expression of Vldlr. Similarly, exposure of human Huh-7 hepatocytes to GW501516 increased the expression of VLDLR and triglyceride accumulation, the latter being prevented by VLDLR knockdown. Finally, treatment with another PPARβ/δ agonist increased VLDLR levels in the liver of wild-type mice, but not PPARβ/δ-deficient mice, confirming the regulation of hepatic VLDLR by this nuclear receptor. Our results suggest that upregulation of hepatic VLDLR by PPARβ/δ agonists might contribute to the hypolipidemic effect of these drugs by increasing lipoprotein delivery to the liver. Overall, these findings provide new effects by which PPARβ/δ regulate VLDLR levels and may influence serum triglyceride levels and NAFLD development.
El receptor de las lipoproteínas de muy baja densidad (VLDLR) desempeña una función muy importante en el control de los niveles de triglicéridos séricos y en el desarrollo de la enfermedad del hígado graso no alcohólico (EHGNA). En este estudio hemos investigado el papel de la activación del receptor activado por los proliferadores peroxisómicos (PPAR)β/δ en la regulación hepática del VLDLR. El tratamiento de ratones alimentados con una dieta rica en grasas con el agonista PPARβ/δ GW501516 aumentó la expresión hepática de Vldlr. Asimismo, la exposición de hepatocitos humanos Huh-7 a GW501516 aumentó la expresión de VLDLR y la acumulación de triglicéridos, siendo este ultimo aumento evitado por el knockdown de VLDLR. Finalmente, el tratamiento con otro agonista PPARβ/δ incrementó los niveles de VLDLR en el hígado de ratones wild-type, pero no en el de ratones deficientes en PPARβ/δ, confirmando la regulación del VLDLR hepático por este receptor. En conjunto, nuestros resultados proporcionan un nuevo efecto por el que PPARβ/δ regula los niveles de VLDLR y puede influenciar los niveles de triglicéridos séricos así como el desarrollo de la EHGNA.
Nonalcoholic fatty liver disease (NAFLD) is increasingly recognized as the most common chronic liver disease, affecting 25% of the global population.1 NAFLD encompasses a spectrum of liver injuries ranging from hepatic steatosis, defined by the excessive accumulation of triglycerides in the liver, to the most severe condition of non-alcoholic steatohepatitis (NASH), which is characterized by hepatic steatosis, inflammation and damage of liver cells. In addition, NAFLD is an important risk factor for the development of obesity-related pathologies including type 2 diabetes mellitus (T2DM) and cardiovascular diseases.2 Patients suffering NAFLD also show dyslipidemia, which is characterized by increased levels of serum triglycerides and decreased levels of HDL cholesterol.3–5 The mechanisms responsible for the profound alterations in serum and liver triglyceride levels in NAFLD are not well understood, but they might involve changes in the delivery of the main circulating particles transporting these lipids, the very low-density lipoprotein (VLDL).
In the last years it has been demonstrated that VLDL receptor (VLDLR) plays an important role in the development of hepatic steatosis and in controlling the levels of serum triglycerides.6 This receptor belongs to the low-density lipoprotein (LDL) receptor family and its expression is high in brain, heart, skeletal muscle, and adipose tissue, whereas its very low in the liver under physiological conditions.7,8 VLDLR binds apolipoprotein E (apoE) triglyceride-rich lipoproteins such as chylomicrons, VLDL, and intermediate density lipoproteins, leading to lipid entry into the cell through lipoprotein lipase (LPL)-dependent lipolysis or receptor-mediated endocytosis.9–12 As a result, a link has been established between VLDLR levels and plasma triglyceride levels.13 Mice lacking VLDLR are leaner, display normal blood lipids14 and are protected from obesity induced by high-fat diet (HFD) feeding.15 However, fasting or feeding a HFD increases serum triglyceride levels in these animals.15,16 VLDLR is regulated by several transcription factors, including peroxisome proliferator-activated receptor (PPAR)γ in adipose tissue12 and hypoxia-inducible factor 1α (HIF-1α) in the heart,17 contributing to lipid deposition in both tissues. In alcoholic liver disease, the activation of oxidative stress-induced nuclear factor (erythroid-derived 2)-like 2 (Nrf2) upregulates VLDLR levels,18 whereas stimulation of activating transcription factor 4 (ATF4) signaling during endoplasmic reticulum stress induces hepatic steatosis via increase in VLDLR levels by enhancing lipoprotein delivery to the liver.6 In addition, hepatic VLDLR upregulation plays an essential role in the triglyceride-lowering effect of fenofibrate through PPARα activation.19 However, little is known about the effects of PPARβ/δ on VLDLR regulation in the liver. PPARβ/δ is a ligand-activated transcription factor involved in the regulation of glucose and lipid homeostasis,20 and it has been proposed as a therapeutic target for the treatment of metabolic syndrome.21 Thus, genetic manipulation of PPARβ/δ as well as its activation by agonists attenuate dyslipidemia and hyperglycemia, improve whole-body insulin sensitivity, and prevent diet-induced obesity.22 In this study, we demonstrate that PPARβ/δ activation upregulates VLDLR levels in liver, thereby increasing VLDL delivery to the liver, providing a novel mechanism by which PPARβ/δ activation can ameliorate hypertriglyceridemia.
Materials and methodsReagentsControl and VLDLR siRNA were purchased from Santa Cruz (Dallas, TX). Triglyceride levels were measured using a commercial kit (Sigma, St. Louis, MO).
Plasma VLDL isolationVLDL particles (d<1.006g/mL) were isolated by ultracentrifugation from human plasma of healthy subjects (total cholesterol ≤5.2mmol/L, triglyceride ≤1mmol/L) obtained in EDTA-containing vacutainer tubes. The VLDL preparation was extensively dialyzed in PBS and then triglyceride concentration was measured using a commercial kit adapted to a COBAS c501 autoanalyzer (Roche Diagnostics, Rotkreuz, Switzerland). Cells were treated with 300μg/mL of filtered VLDL, based on triglyceride concentration, as previously described.23
MiceMale CD1 mice (Harlan Ibérica S.A., Barcelona, Spain) were randomly distributed into three experimental groups (n=8 each): standard diet, Western-type high-fat diet (HFD, 35% fat by weight, 58% Kcal from fat, Harlan Ibérica S.A.) plus one daily oral gavage of vehicle (0.5%, w/v, carboxymethylcellulose), and HFD plus one daily oral dose of 3mgkg−1day−1 of the PPARβ/δ agonist GW501516 dissolved in the vehicle (volume administered 1mLkg−1) for 3 weeks.
Male Pparβ/δ knockout (Pparβ/δ−/−) mice and their wild-type littermates (Pparβ/δ+/+) with the same genetic background (C57BL/6X129/SV)24 and an initial weight of 20–25g were fed a standard diet. When indicated, wild-type and Pparβ/δ-null mice were treated with daily intragastric doses of the PPARβ/δ agonist GW0742 (10mgkg−1day−1) or a vehicle solution (1.0% w/v carboxymethylcellulose) for 14 days.
The research complied with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). All procedures were approved by the University of Barcelona Bioethics Committee, as stated in Law 5/21 July 1995 passed by the Generalitat de Catalunya.
Cell cultureHuman Huh-7 cells (a kind gift from Dr. Mayka Sanchez from Josep Carreras Leukemia Research Institute) were cultured in DMEM supplemented with 10% serum, at 37°C/5% CO2. siRNA transfections were performed with Lipofectamine 2000 (Life Technologies) following the protocol provided by the manufacturer.
RNA preparation and quantitative RT-PCRThe relative levels of specific mRNAs were assessed by real-time RT-PCR, as previously described.25 Primer sequences used for real-time RT-PCR are displayed in Table 1.
Primer sequences used for real-time RT-PCR.
| Gene | Primers | |
|---|---|---|
| mAngplt4 | for | 5′-GCATGGCTGCCTGTGGTAAC-3′ |
| rev | 5′-ATCTTGCTGTTTTGAGCCTTGA-3′ | |
| mAprt | for | 5′-CAGCGGCAAGATCGACTACA-3′ |
| rev | 5′-AGCTAGGGAAGGGCCAAACA-3′ | |
| mPdk4 | for | 5′-CACCACATGCTCTTCGAACTCT-3′ |
| rev | 5′-AAGGAAGGACGGTTTTCTTGATG-3′ | |
| hVLDLR | for | 5′-CAAGAGGAAGTTCCTGTTTAACTCTGA-3′ |
| rev | 5′-TGACCAGTAAACAAAGCCAGACA-3′ | |
| mVldlr | for | 5′-TCCAATGGCCTAATGGAATTACA-3′ |
| rev | 5′-AGCATGTGCAACTTGGAATCC-3′ |
Isolation of total protein extracts was performed as described elsewhere.25 Proteins (30μg) were separated by SDS-PAGE on 10% acrylamide separation gels and transferred to Immobilon polyvinylidene difluoride membranes (Millipore). Western blot analysis was performed using antibodies against VLDLR (AF2258) (R&D Systems, Minneapolis, MN) and actin (A5441) (Sigma-Aldrich, Madrid, Spain). Detection was achieved using the Western Lightning® Plus-ECL chemiluminescence kit (PerkinElmer, Waltham, MA). The equal loading of proteins was assessed by Ponceau S staining. The size of detected proteins was estimated using protein molecular-mass standards (Bio-Rad).
Hematoxylin–eosin and Oil Red O stainingWe performed hematoxylin–eosin and Oil Red O staining as previously reported.25 ORO staining was quantified using Image J software.
Statistical analysesResults are expressed as means±S.D. Significant differences were established by two-way ANOVA using the GraphPad Instat program (GraphPad Software V5.01) (GraphPad Software Inc., San Diego, CA). When significant variations were found by two-way ANOVA, the Tukey–Kramer multiple comparison post-test was performed. Differences were considered significant at p<0.05.
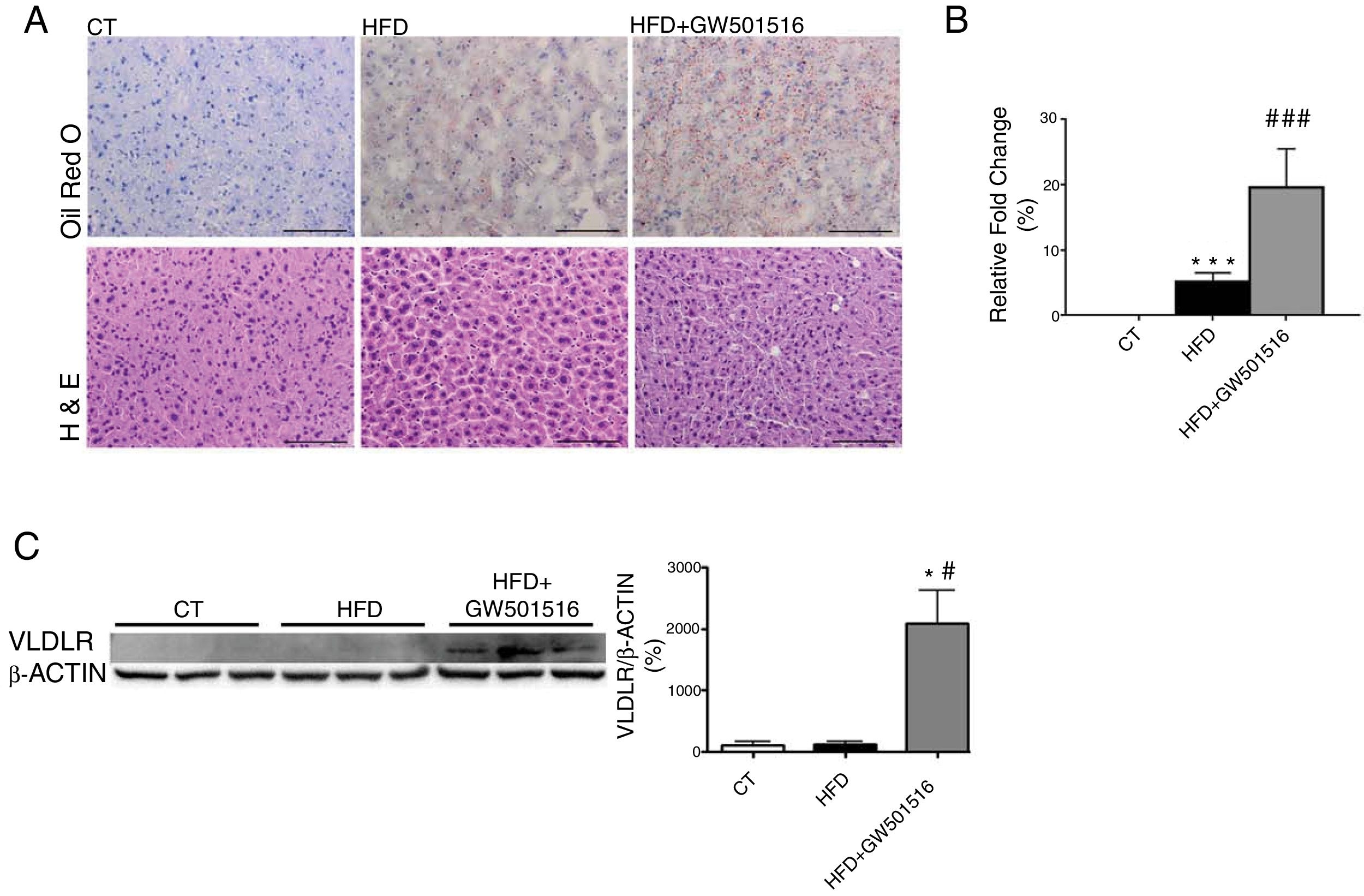
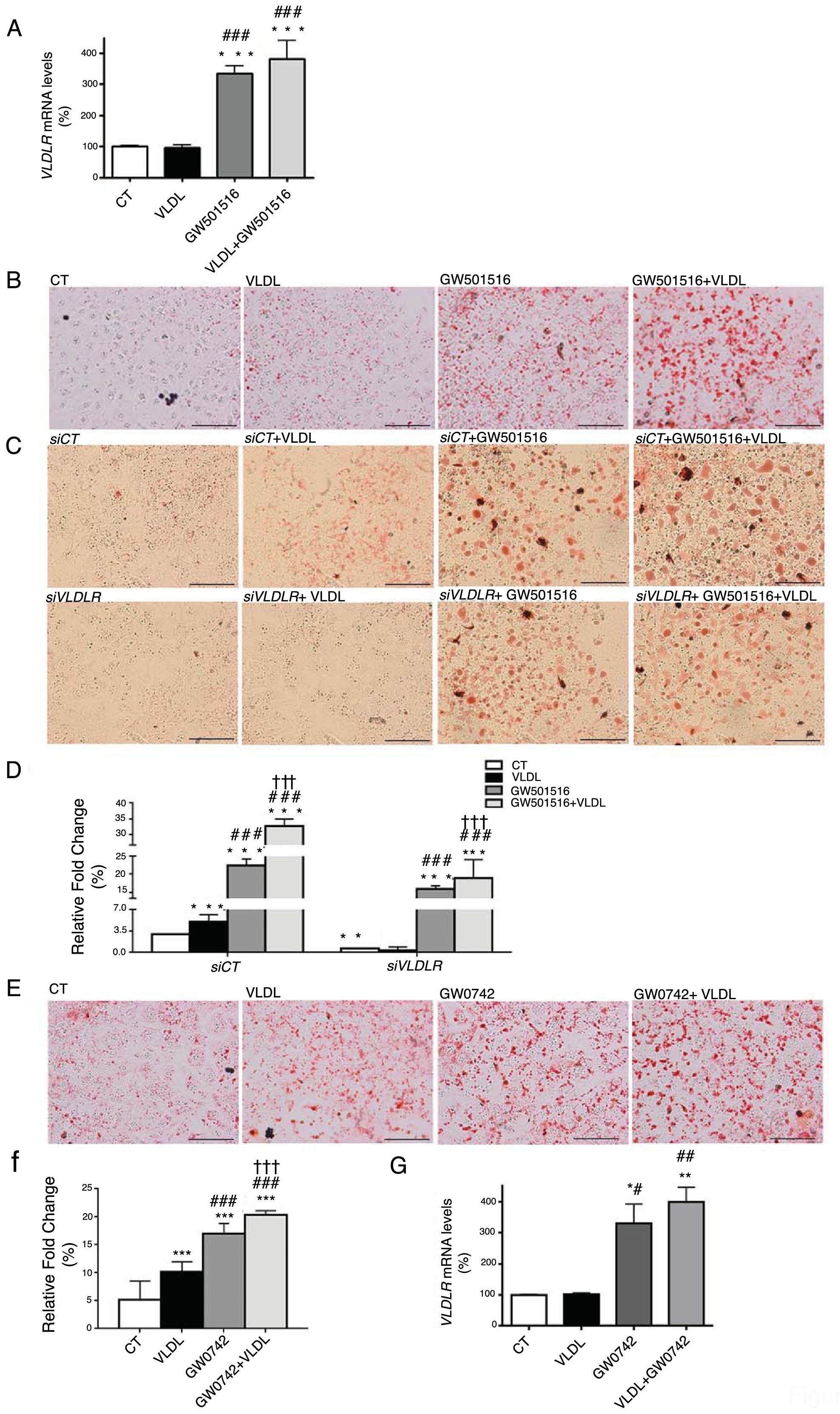
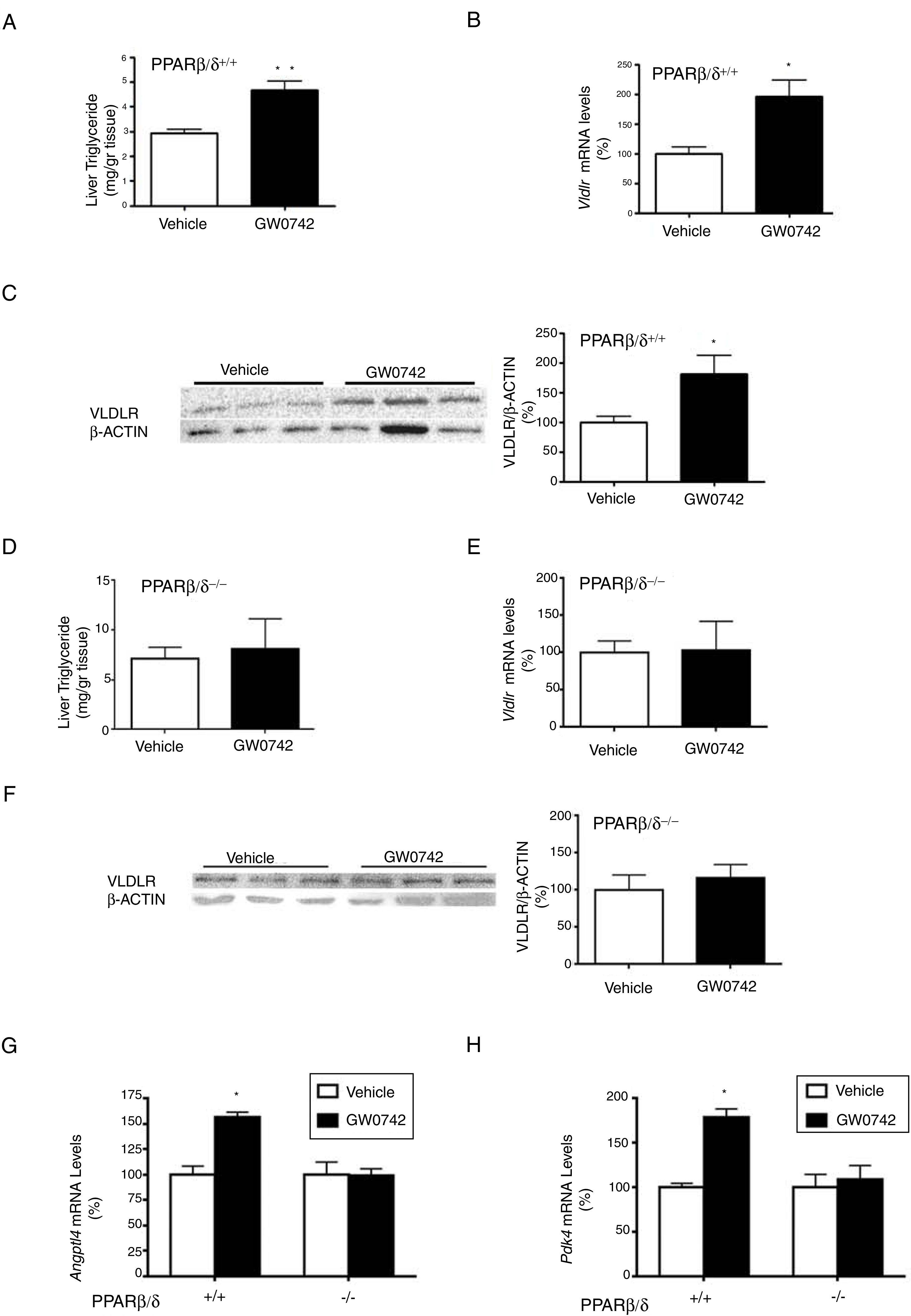
ResultsWe have previously reported that administration of the PPARβ/δ activator GW501516 to mice fed a HFD for a short period (3 weeks) reduced plasma triglyceride levels.26 However, the hypotriglyceridemic effect observed at 3 weeks in these mice26 was not accompanied by a reduction in hepatic steatosis. In fact, HFD-fed mice receiving GW501516 showed higher hepatic lipid levels than those treated with vehicle, as assessed by Oil Red O (ORO) and hematoxylin–eosin staining (Fig. 1A and B). This finding suggests that the hypotriglyceridemic effect of PPARβ/δ activators observed at 3 weeks may be mediated only in part by an increase in fatty acid oxidation,26 although fatty acid oxidation may be the main contributor in longer treatments (8 weeks), where hepatic steatosis is not observed any more following treatment with GW501516.27 This prompted us to examine whether the hypotriglyceridemic effect caused by GW501516 at 3 weeks was mediated by increasing lipoprotein delivery to the liver through VLDLR. Interestingly, VLDLR protein levels (Fig. 1C) were increased only in the livers of mice treated with GW501516. Similarly, incubation of human Huh-7 hepatocytes with GW501516 also increased the expression of VLDLR either in the absence or in the presence of human VLDL (Fig. 2A). Next, human Huh-7 hepatocytes, which secrete VLDL, although in low amounts,28 were incubated with human VLDL, GW510156 or both. Incubation with VLDL or GW501516 led to some neutral lipid accumulation, especially with the PPARβ/δ agonist (Fig. 2B), where a strong increase was observed in VLDLR mRNA levels (Fig. 2A), thus allowing a higher uptake of VLDL lipoproteins secreted by hepatocytes. Interestingly, lipid accumulation was strongly increased when cells were exposed to VLDL and treated with GW501516 (Fig. 2B), suggesting that PPARβ/δ activation upregulates the uptake of these lipoproteins through VLDLR. Similar results were obtained with another PPARβ/δ agonist, GW0742 (Fig. 2D–F). In line with these results, knockdown of VLDLR by transfecting cells with siRNA against this gene reduced VLDL uptake caused by GW501516, suggesting that PPARβ/δ activation upregulates the uptake of these lipoproteins through VLDLR (Fig. 2B and C). When wild-type and Pparβ/δ-deficient mice were treated with the PPARβ/δ agonist GW0742, we observed that this drug increased hepatic liver triglyceride and the hepatic expression and protein levels of VLDLR in the liver of wild-type, but not of Pparβ/δ-null mice (Fig. 3A–F). The expression of two well-known PPARβ/δ-target genes (Angptl4 and Pdk4) showed the same behavior (Fig. 3G–H). These findings confirm that the effects of PPARβ/δ activators on VLDLR were dependent on PPARβ/δ.
. Animals were sacrificed after three weeks of treatment. (A) Oil Red O and hematoxylin–eosin (H&E) staining of livers. Scale bar: 100μm. (B) Quantification of ORO staining of Fig. 1A. Data are presented as the mean±S.D. (n=4 per group). (C) Liver cell lysates were assayed for Western blot analysis with antibody against VLDLR. *p<0.05 vs. WT mice fed a standard diet (CT). #p<0.05 vs. mice fed a HFD. Data are presented as the mean±S.D. (n=5 per group) relative to the control (CT) group. ***p<0.001 and *p<0.05 vs. CT group. ###p<0.001 and #p<0.05 vs. mice fed the HFD.")
PPARβ/δ activators increase VLDLR abundance in liver. Male mice were fed a standard chow or HFD with or without GW501516 (3mgkg−1day−1). Animals were sacrificed after three weeks of treatment. (A) Oil Red O and hematoxylin–eosin (H&E) staining of livers. Scale bar: 100μm. (B) Quantification of ORO staining of Fig. 1A. Data are presented as the mean±S.D. (n=4 per group). (C) Liver cell lysates were assayed for Western blot analysis with antibody against VLDLR. *p<0.05 vs. WT mice fed a standard diet (CT). #p<0.05 vs. mice fed a HFD. Data are presented as the mean±S.D. (n=5 per group) relative to the control (CT) group. ***p<0.001 and *p<0.05 vs. CT group. ###p<0.001 and #p<0.05 vs. mice fed the HFD.
 mRNA abundance of VLDLR. (B) Oil Red O staining in Huh-7 hepatocytes incubated for 24h in the absence (Control, CT) or presence of VLDL (300μg/mL) and/or GW501516 (10μM). (C) Oil Red O staining in Huh-7 hepatocytes non-transfected or transfected with siRNA control or siRNA against VLDLR and incubated for 24h in the absence (Control, CT) or presence of VLDL (300μg/mL) and/or GW501516 (10μM). Scale bar: 100μm. (D) Quantification of ORO staining of Fig. 2B. Data are presented as the mean±S.D. (n=4 per group). (E) Oil Red O staining in Huh-7 hepatocytes incubated for 24h in the absence (Control, CT) or presence of VLDL (300μg/mL) and/or GW0742 (10μM). (F) Quantification of ORO staining of Fig. 2D. Data are presented as the mean±S.D. (n=4 per group). (G) mRNA abundance of VLDLR. ***p<0.001, **p<0.01 and *p<0.05 vs. CT group. ###p<0.001, ##p<0.01 and #p<0.05 vs. VLDL-exposed cells. †††p<0.001 vs. GW501516-exposed cells.")
Intracellular accumulation of triglycerides in hepatocytes caused by GW501516 is inhibited by VLDLR knockdown. (A) mRNA abundance of VLDLR. (B) Oil Red O staining in Huh-7 hepatocytes incubated for 24h in the absence (Control, CT) or presence of VLDL (300μg/mL) and/or GW501516 (10μM). (C) Oil Red O staining in Huh-7 hepatocytes non-transfected or transfected with siRNA control or siRNA against VLDLR and incubated for 24h in the absence (Control, CT) or presence of VLDL (300μg/mL) and/or GW501516 (10μM). Scale bar: 100μm. (D) Quantification of ORO staining of Fig. 2B. Data are presented as the mean±S.D. (n=4 per group). (E) Oil Red O staining in Huh-7 hepatocytes incubated for 24h in the absence (Control, CT) or presence of VLDL (300μg/mL) and/or GW0742 (10μM). (F) Quantification of ORO staining of Fig. 2D. Data are presented as the mean±S.D. (n=4 per group). (G) mRNA abundance of VLDLR. ***p<0.001, **p<0.01 and *p<0.05 vs. CT group. ###p<0.001, ##p<0.01 and #p<0.05 vs. VLDL-exposed cells. †††p<0.001 vs. GW501516-exposed cells.
 and Pparβ/δ-null mice were treated with vehicle or GW0742 (10mgkg−1day−1) for two weeks (n=6 per group) and liver triglyceride levels (A and E), hepatic mRNA abundance (B and E), protein levels (C and F) of VLDLR and the expression of hepatic Angptl4 (G) and Pdk4 (H) were analyzed. **p<0.01 and *p<0.05 vs. wild-type mice treated with vehicle.")
The effect of GW0742 on VLDLR in liver is PPARβ/δ-dependent. Wild-type (WT) and Pparβ/δ-null mice were treated with vehicle or GW0742 (10mgkg−1day−1) for two weeks (n=6 per group) and liver triglyceride levels (A and E), hepatic mRNA abundance (B and E), protein levels (C and F) of VLDLR and the expression of hepatic Angptl4 (G) and Pdk4 (H) were analyzed. **p<0.01 and *p<0.05 vs. wild-type mice treated with vehicle.
Here we present evidence that pharmacological activation of PPARβ/δ upregulates VLDLR in liver, and this effect might contribute to the hypotriglyceridemic action of PPARβ/δ activators by increasing lipoprotein delivery to the liver. As a result, PPARβ/δ activation increases intrahepatic triglyceride accumulation in short treatments (3–4 weeks) (this manuscript and 30), but the activation of fatty acid oxidation by PPARβ/δ agonists in longer treatments (8 weeks)26 finally reverts hepatic triglyceride accumulation.27 Thus, in addition to PPARα activators, such as fenofibrate, which require the upregulation of hepatic VLDLR via PPARα for their hypotriglyceridemic effect,19 VLDLR may play a key role in the triglyceride-lowering effect of PPARβ/δ agonists. Our findings also demonstrate that the effect of these compounds depends on PPARβ/δ because it is abolished in Pparβ/δ-deficient mice. A functional peroxisome-proliferator response element (PPRE) for PPARα has been described in the Vldlr promoter region,19 suggesting that, as with many PPARα-responsive genes, this PPRE also responds to PPARβ/δ. The effect of PPARα on VLDLR seems to be tissue-specific, since fenofibrate increases VLDLR expression in hepatic cells but not in adipose cells.19 Likewise, PPARβ/δ regulation of the Vldlr gene may also be tissue-specific, since it has been reported that PPARβ/δ activation induces microRNA-100 and decreases VLDLR expression in endothelial cells.28
In spite of the upregulation of VLDLR by PPARβ/δ activators, we have previously reported that Pparβ/δ-null mice showed increased hepatic VLDLR levels through indirect activation of ATF4 and Nrf2,29 another transcription factor that contributes to increase VLDLR levels.18 Under conditions of Pparβ/δ-deficiency, the increase in VLDLR levels would contribute to hepatic steatosis, since in contrast to what it is observed following PPARβ/δ activation, where hepatic fatty acid oxidation has been reported to increase,26 the lack of increase in fatty acid oxidation would exacerbate triglyceride accumulation. In addition, stimulation of ER stress induces hepatic steatosis through VLDLR,6 and this process might be exacerbated in Pparβ/δ-null mice through a higher activation of both ATF4 and Nrf2, suggesting that the absence of PPARβ/δ contributes to intensify ER stress-induced fatty liver.
Although it might seem contradictory that mice lacking VLDLR display normal lipids14 with the results obtained in this study, it is worth saying that this receptor is widely expressed in the heart, skeletal muscles, adipose tissues, and macrophages, whereas it is barely detectable in the liver under normal conditions.7,8 Interestingly, the hepatic expression of VLDLR is upregulated by fenofibrate and this increase was correlated with a reduction in serum triglycerides, whereas the absence of VLDLR blunted the hypotriglyceridemic effect of the fibrate.19 These findings suggest that the increase in the hepatic levels of VLDLR is essential for serum triglyceride metabolism.
In summary, the findings of this study suggest that the hypotriglyceridemic effect of PPARβ/δ activators might involve an increase in hepatic VLDLR levels. Overall, these data suggest that modulation of VLDLR abundance by PPARβ/δ agonists might contribute to the hypolipidemic effect of these drugs by increasing lipoprotein delivery to the liver.
Author's contributionMZ, EB, XP, JCEG, LC, and MVC performed the experiments; MZ, WW and MVC designed the experiments and revised the results; MVC was primarily responsible for writing the manuscript. All authors contributed to manuscript editing and approval.
Financial supportThis study was partly supported by funds from the SEA-FEA grant for basic research 2017 and the Spanish Ministry of the Economy and Competitiveness (SAF2015-64146-R to MVC) and European Union ERDF funds. CIBER de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM) is a Carlos III Health Institute projects. WW is supported by Start-Up Grants from the Lee Kong Chian School of Medicine, Nanyang Technological University, Singapore, and by the Région Midi-Pyrénées, France.
Conflict of interestThe authors declare no competing interests.