






Epidemiologic evidence supported an inverse association between HDL (high-density lipoprotein) cholesterol (HDL-C) levels and atherosclerotic cardiovascular disease (ASCVD), identifying HDL-C as a major cardiovascular risk factor and postulating diverse HDL vascular- and cardioprotective functions beyond their ability to drive reverse cholesterol transport. However, the failure of several clinical trials aimed at increasing HDL-C in patients with overt cardiovascular disease brought into question whether increasing the cholesterol cargo of HDL was an effective strategy to enhance their protective properties. In parallel, substantial evidence supports that HDLs are complex and heterogeneous particles whose composition is essential for maintaining their protective functions, subsequently strengthening the “HDL quality over quantity” hypothesis.
The following state-of-the-art review covers the latest understanding as per the roles of HDL in ASCVD, delves into recent advances in understanding the complexity of HDL particle composition, including proteins, lipids and other HDL-transported components and discusses on the clinical outcomes after the administration of HDL-C raising drugs with particular attention to CETP (cholesteryl ester transfer protein) inhibitors.
Estudios epidemiológicos respaldan una asociación inversa entre los niveles de colesterol de lipoproteínas de alta densidad (c-HDL) y la enfermedad cardiovascular aterosclerótica (ECVA), identificando el c-HDL como un importante factor de riesgo cardiovascular y postulando diversas funciones vasculares y cardioprotectoras de las HDL más allá de su capacidad para promover el transporte reverso del colesterol. Sin embargo, el fracaso de varios ensayos clínicos dirigidos a aumentar el c-HDL en pacientes con enfermedad cardiovascular manifiesta, puso en duda el concepto que incrementar la carga de c-HDL fuera una estrategia eficaz para potenciar sus propiedades protectoras. Paralelamente, numerosos estudios han evidenciado que las HDL son partículas complejas y heterogéneas cuya composición es esencial para mantener sus funciones protectoras, lo que refuerza la hipótesis de que «la calidad de las HDL prima sobre la cantidad».
En el siguiente manuscrito revisamos el estado del arte sobre los últimos avances en torno a las funciones de las HDL en la ECVA, nos adentramos en los avances recientes en la comprensión de la complejidad de la composición de las partículas de HDL, incluidas las proteínas, los lípidos y otros componentes transportados por las HDL, y revisamos los resultados clínicos tras la administración de inductores del c-HDL, especialmente los inhibidores de la proteína transportadora del colesterol esterificado (CETP).
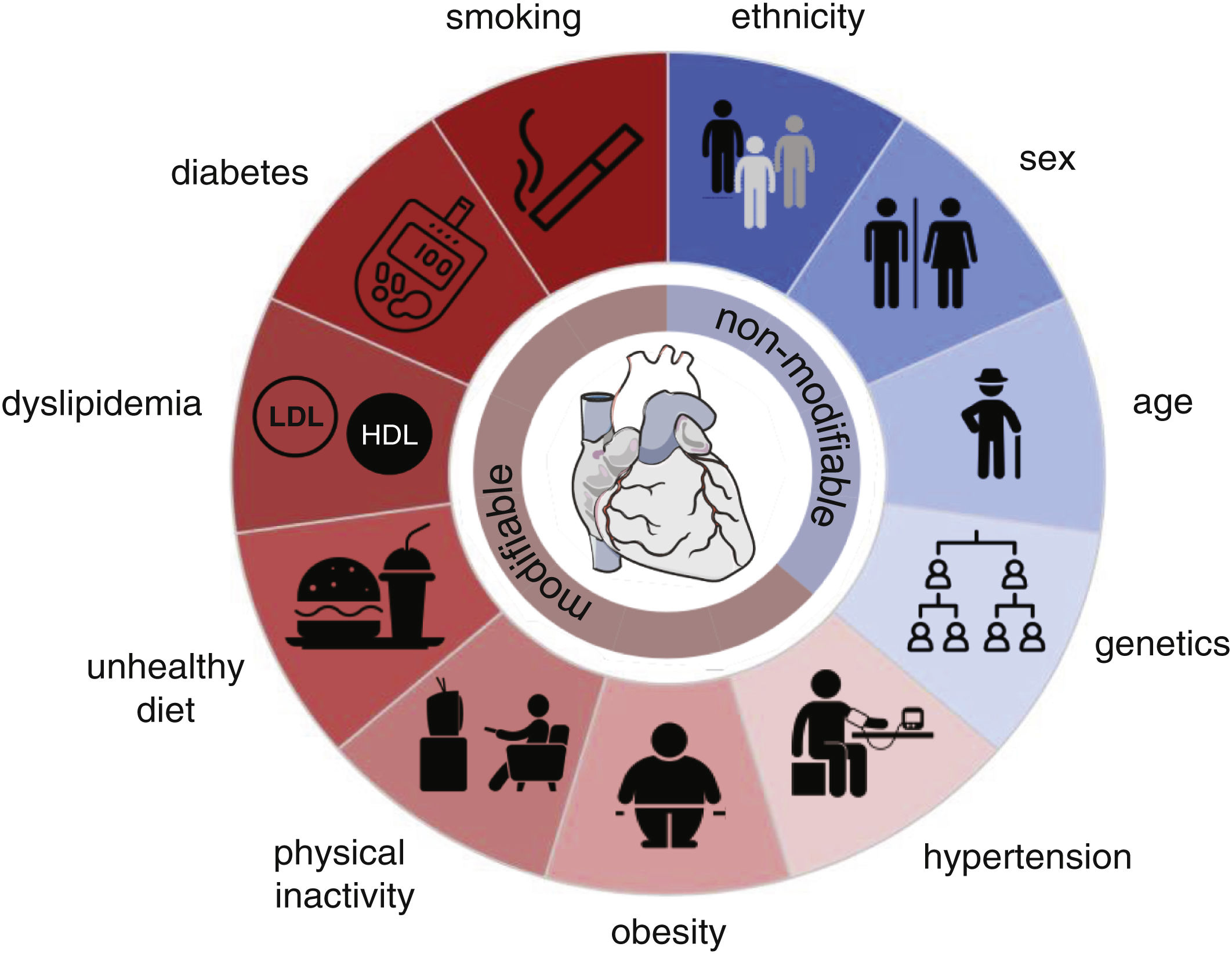
Despite recent advances in preventing, detecting, and treating cardiovascular disease (CVDs), the global burden is increasing due to population growth and ageing. Within the last 30 years, cardiovascular (CV) mortality has risen by 53%,1 remaining the leading cause of death worldwide, accounting for 32% of all deaths, of which the two most prevalent causes of death are ischemic heart disease (16%) and stroke (12%).2 The probability of suffering from any CVD increases with risk factors (Fig. 1).
 and non-modifiable (blue) risk factors. This figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license (heart), and The Noun Project (symbols). Donut chart by google docs (excel).")
Risk factors for atherosclerotic cardiovascular diseases. Modifiable (red) and non-modifiable (blue) risk factors. This figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license (heart), and The Noun Project (symbols). Donut chart by google docs (excel).
As a largely asymptomatic condition, elevated blood pressure is estimated to be responsible for around 50% of all deaths from ischemic heart disease and stroke.3 It is thus considered the single largest contributor to the global burden of disease and mortality. Being overweight (BMI≥25kg/m2) and subsequently obese (BMI≥30kg/m2) are continuously on the rise and have become a global health crisis due to their increased risk of suffering from numerous diseases, including CVD. Directly linked to obesity are physical inactivity and an unhealthy and unbalanced diet with high amounts of sodium (>5g/day), saturated fat, cholesterol, and red or processed meat but low intake of fruits, vegetables, whole grains, legumes, fibre, nuts, seeds, omega-3 and polyunsaturated fats which increase CV risk and thus contribute to overall CV deaths.1,4 Consequently, these dietary habits can lead to dyslipidaemia, an imbalance of serum lipids. As such, high total cholesterol, low-density lipoprotein cholesterol (LDL-C), triglyceride levels, and lower high-density lipoprotein cholesterol (HDL-C) concentrations are strongly associated with the development of atherosclerotic CVDs (ACVDs). Furthermore, besides developing a proatherogenic profile,5 serum lipid levels may induce a prothrombotic state.6 Likewise, lifestyle-related hyperglycaemia and diabetes mellitus are increasing problems since age-standardized mean fasting plasma glucose levels have continuously risen during the past 30 years, regardless of sex, age, and country.7,8 Compared to 1990, there has been an increase in diabetes of over 186%.7 Finally, smoking is one of the more difficult-to-grasp risk factors due to the complexity of the chemical constituents of the smoke. Metal components in cigarette smoke have been shown to damage vascular endothelial cells by inducing oxidative stress and inflammation, which are involved in the development of ACVDs progression.9
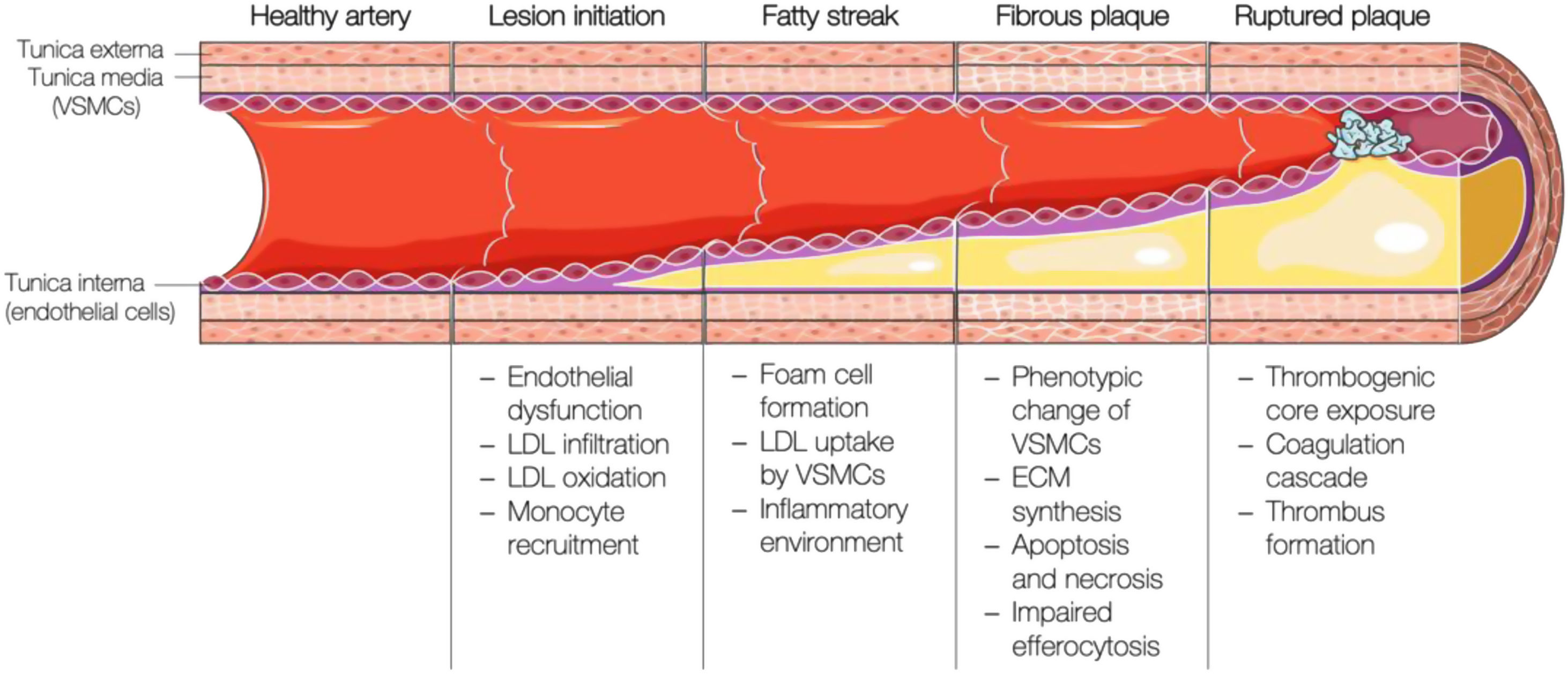
Brief overview of the pathogenesis of atherosclerosisAtherosclerosis is a lipid-driven inflammatory disease characterised by the deposition of fatty, fibrous, and calcified material in the innermost layer of large and medium-sized arteries. Its development starts with the activation of the arterial endothelium, followed by an inflammatory cascade, continuous lipid build-up in the vessel wall, and plaque formation, which ultimately will disrupt and induce thrombosis (i.e., atherothrombosis) with impaired or even blocked blood flow (Fig. 2).

Schematic representation of the different stages of atherosclerotic plaque formation. VSMCs: vascular smooth muscle cells; LDL: low-density lipoprotein; ECM: extracellular matrix. This figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
The first step in the development of atherosclerosis is endothelial activation and dysfunction due to hemodynamic forces within the arteries.10 Lesion-prone regions such as arterial bifurcations are mainly characterised by disturbed laminar flow and reduced shear stress,11,12 which disrupt the endothelial barrier and facilitate the retention of LDL particles,13,14 but also upregulate the expression of genes involved in the attraction and recruitment of immune cells (MCP-1,15 VCAM-1,16 ICAM-1,17 PECAM-118). On the other hand, fluid mechanical forces also affect the expression and activity of the endothelial nitric oxide synthase (eNOS), the enzyme catalysing the conversion from l-arginine to nitric oxide (NO). Endothelium-derived NO production and bioavailability are key for the homeostasis of the vascular endothelium, adjacent smooth muscle cell vasorelaxation, and inhibition of platelet activation, adhesion, and aggregation.19
High levels of LDL-C favour LDL particle infiltration into the arterial intima by diffusion, paracellular crossing, or transcytosis,20 where they suffer modifications. Circulating monocytes are then attracted and extravasate to the intima to engulf these modified LDL particles and become foam cells. Foam cells secrete pro-inflammatory cytokines (mainly interleukin (IL)-1, IL-6, and tumour necrosis factor-alpha (TNFα)) and chemokines (MCP-1), which further promote monocyte recruitment and inflammatory response propagation.21
Due to a cocktail of mainly macrophage-derived growth factors and cytokines, vascular smooth muscle cells (VSMCs) are recruited to the luminal side of the lesion and secrete extracellular matrix components, matrix metalloproteases, and pro-inflammatory cytokines.22 As atherosclerotic plaques mature and more cholesterol is deposited, foam cells and intimal VSMCs cannot cope with the excessive cholesterol burden and undergo apoptosis.23 Cellular debris, cholesterol crystals, and thrombogenic, inflammatory, and oxidative material accumulate in an extending necrotic core. Osteochondrogenic VSMC-derived calcifications gradually extend from the necrotic centre to the surrounding matrix, which, together with increasing ECM production, causes arterial stiffness.24,25 Upon rising apoptosis rates in the intimal VSMC population and matrix metalloprotease activity, the plaque becomes vulnerable,10 which is generally considered to include a large necrotic core, a thin fibrous cap, and an increased inflammatory infiltrate due to continuous exposure to the pro-atherogenic milieu.10 When the fibrous cap that protects circulation from the content of the necrotic core fissures or ruptures, the thrombogenic and pro-inflammatory plaque content is exposed to the blood. Upon contact with blood components (platelets and coagulant factors), sub-occlusive or occlusive thrombosis may occur, leading to acute coronary syndromes.26
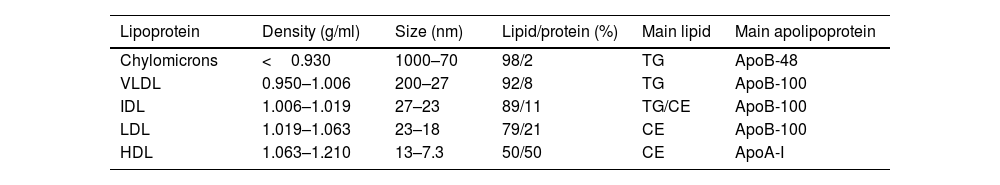
Lipoprotein particlesLipoproteins are complex particles that enable the transportation of non-polar lipids such as triglycerides and cholesteryl esters in their hydrophobic core, protected by an outer hydrophilic layer containing free cholesterol, phospholipids, and apolipoproteins. They primarily differ in size, density, and composition, as summarised in Table 1.
Classification and characteristics of lipoproteins.
| Lipoprotein | Density (g/ml) | Size (nm) | Lipid/protein (%) | Main lipid | Main apolipoprotein |
|---|---|---|---|---|---|
| Chylomicrons | <0.930 | 1000–70 | 98/2 | TG | ApoB-48 |
| VLDL | 0.950–1.006 | 200–27 | 92/8 | TG | ApoB-100 |
| IDL | 1.006–1.019 | 27–23 | 89/11 | TG/CE | ApoB-100 |
| LDL | 1.019–1.063 | 23–18 | 79/21 | CE | ApoB-100 |
| HDL | 1.063–1.210 | 13–7.3 | 50/50 | CE | ApoA-I |
VLDL: very low-density lipoprotein; IDL: intermediate-density lipoprotein; LDL: low-density lipoprotein; HDL: high-density lipoprotein; Lp(a): lipoprotein(a); TG: triglycerides; CE: cholesteryl ester; ApoB: apolipoprotein B; ApoA: apolipoprotein A.
Chylomicrons are the largest and least dense lipoprotein particles, taking dietary triglycerides and cholesterol (exogenous origin from diet) from the intestine to transport to muscle and adipose tissue. The enzymatic action of the lipoprotein lipase at the luminal surface of the adipose and muscle tissues capillary endothelium hydrolyses the triglycerides into glycerol and free fatty acids (lipolysis). Free fatty acids are either used as local energy substrates (beta-oxidation and Krebs cycle) or for energy storage in their re-esterified form (triglycerides). The remaining chylomicron remnants are taken up by hepatic remnant receptors or the LDL receptor (LDLr), and the endogenous lipoprotein pathway is initiated by forming very low-density lipoproteins (VLDLs). VLDLs mainly carry endogenous triglycerides, which either have been synthesised by the liver de novo or produced by the re-esterification of free fatty acids. VLDL-transported triglycerides are hydrolysed at peripheral tissues by lipoprotein lipase, forming VLDL remnants or intermediate-density lipoproteins (IDLs). IDLs are transformed to low-density lipoproteins (LDLs) by further active removal of triglycerides mediated by the cholesteryl ester transport protein (CETP) (to high-density lipoproteins (HDL)) and hepatic triglyceride lipase (to the liver). LDLs are the main cholesterol carrier in circulation, provided for cellular needs by LDLr-mediated uptake and lysosomal digestion of the LDL particle. Excess of apolipoprotein (Apo)-B-containing lipoproteins are considered pro-atherogenic, while the ApoA-containing lipoprotein (HDL particle) may exert anti-atherogenic effects as expanded below.
Classification of the HDL particleThe HDL particle is the densest and smallest of the lipoprotein particle family. They are a group of very heterogeneous particles, and their classification according to physicochemical properties is challenging due to the need for more consensus regarding the definitive categories of HDL subclasses and how to define them.27 The commonly used subclasses are determined according to their separation method (Table 2).
Subclassification of HDL particles.
| Classification | Method | HDL Subclass |
|---|---|---|
| Shape and charge | 2D gel electrophoresis | Pre-ß HDL (discoidal)α-HDL 1/2/3/4 (spherical) |
| Size (nm) | Gradient gel electrophoresis | HDL2a (13.0–9.7), HDL2b (9.7–8.8)HDL3a (8.8–8.2), HDL3b (8.2–7.8), HDL3c (7.8–7.3) |
| Size (nm) | NMR spectroscopy | Large (13–8.8nm)Medium (8.8–8.2nm)Small (8.2–7.3nm) |
| Density (g/ml) | Density gradient ultracentrifugation | HDL2 (1.063–1.125)HDL3 (1.125–1.210) |
HDL: high-density lipoprotein; NMR: nuclear magnetic resonance.
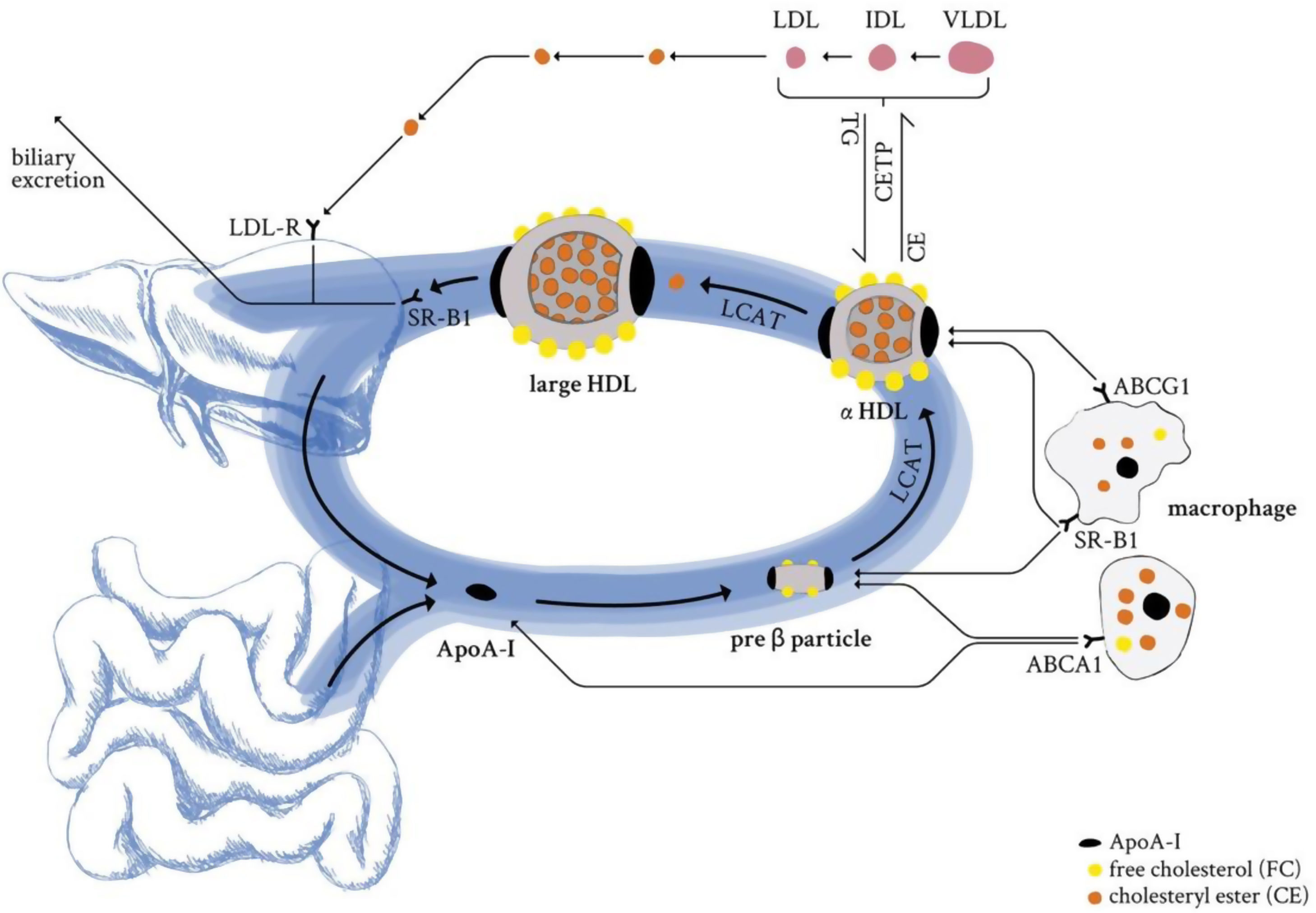
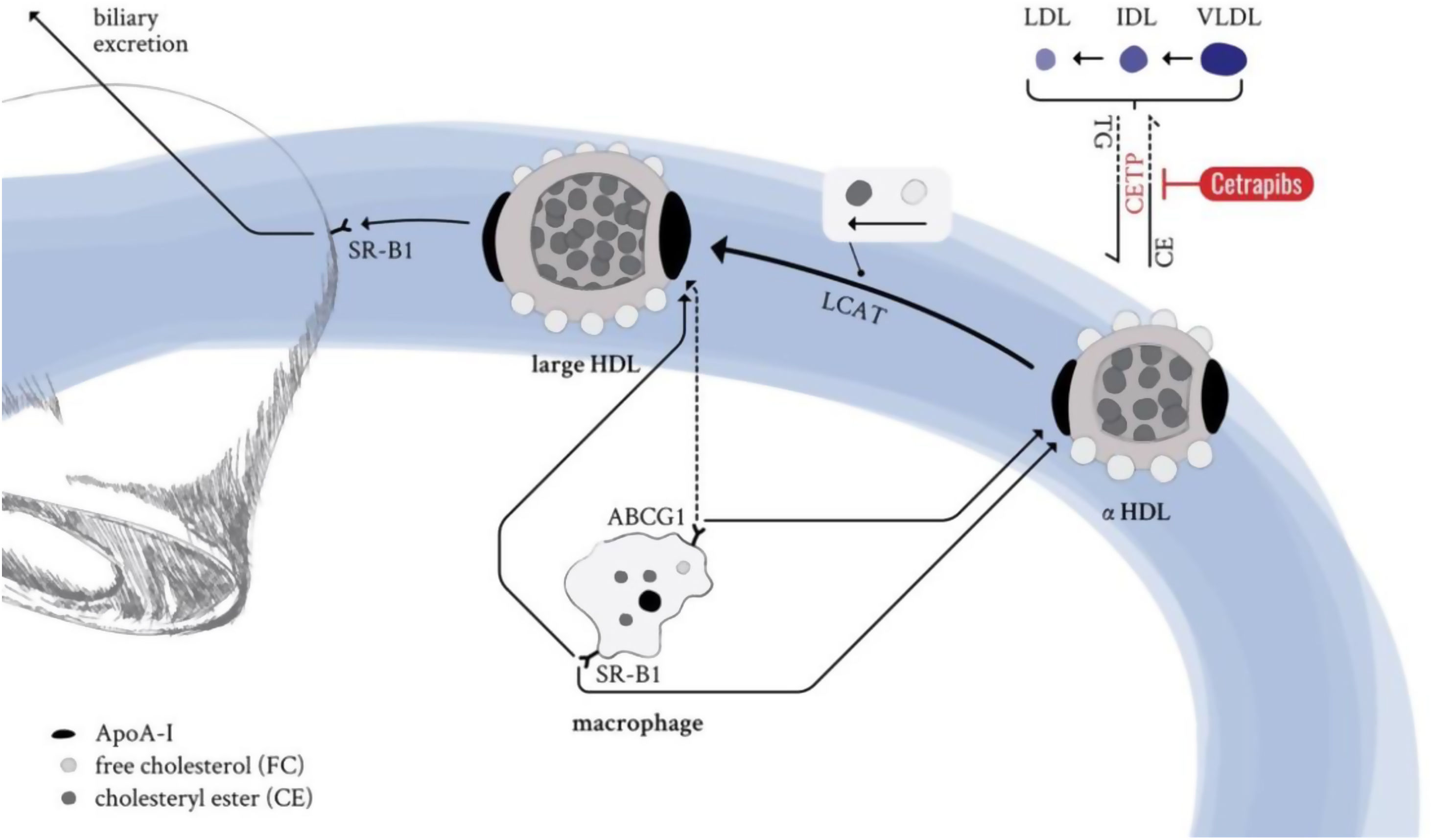
The backbone of all HDL particles is their characteristic structural ApoA-I, synthesised in the liver and intestine. ApoA-I molecules are lipidated by the interaction with ABCA1, which forms nascent discoidal pre-β particles (Fig. 3). Further lipidation and transformation of free cholesterol into cholesteryl ester (by lecithin: cholesterol acyl transferase; LCAT) promote particle maturation to spherical α-HDLs, which primarily interact with ABCG1 and SR-B1 instead of ABCA1 (Fig. 3).
. Further lipidation and conversion of free cholesterol to cholesteryl esters cause particle maturation which is accompanied with an increase in size and compositional complexity. LDL-R: low-density lipoprotein receptor; SR-B1: scavenger receptor B1; LCAT: lecithin cholesterol acyltransferase; CETP: cholesteryl ester transfer protein; TG: triglycerides; CE: cholesteryl ester; ABCG1: ATP binding cassette subfamily G member 1; ABCA1: ATP binding cassette subfamily A member 1; ApoA-I: apolipoprotein A-I; HDL: high-density lipoprotein; LDL: low-density lipoprotein; IDL: intermediate-density lipoprotein; VLDL: very low-density lipoprotein.")
HDL life cycle in healthy conditions. ApoA-I from liver and the intestine is secreted into circulation and loaded with lipids by the interaction with the ABCA1 transporter (pre-β HDL). Further lipidation and conversion of free cholesterol to cholesteryl esters cause particle maturation which is accompanied with an increase in size and compositional complexity. LDL-R: low-density lipoprotein receptor; SR-B1: scavenger receptor B1; LCAT: lecithin cholesterol acyltransferase; CETP: cholesteryl ester transfer protein; TG: triglycerides; CE: cholesteryl ester; ABCG1: ATP binding cassette subfamily G member 1; ABCA1: ATP binding cassette subfamily A member 1; ApoA-I: apolipoprotein A-I; HDL: high-density lipoprotein; LDL: low-density lipoprotein; IDL: intermediate-density lipoprotein; VLDL: very low-density lipoprotein.
This, in combination with the enzymatic activity of CETP or the hepatic and endothelial lipase, as well as the phospholipid transfer activity of the phospholipid transfer protein (PLTP) leads to constant dynamic remodelling of the mature α-HDL particle, thus significantly modulating size and composition. The HDL particle's most important hallmark function is to clear excess cholesterol from peripheral organs and transport it to the liver for biliary excretion (reverse cholesterol transport). HDL-transported cholesteryl esters can either be delivered directly to the hepatocytes (SR-B1) or might be indirectly provided by ApoB-containing lipoproteins that receive HDL-transported cholesteryl esters in exchange for triglycerides. Cholesterol is an essential component of cell membranes and is required for the biosynthesis of vital, biologically active components such as vitamin D, lipid-digesting bile acids, and steroid hormones. Its levels are modulated by the absorption of exogenous, dietary cholesterol and endogenous biosynthesis of cholesterol, mainly performed by the liver and intestine. However, too high circulating cholesterol levels are the underlying cause and the first step towards ACVDs development.
HDL particle compositionHDL particles comprise a lipid core mainly containing the neutral lipids cholesteryl ester and triglycerides, which is protected by a surface layer of amphiphilic phospholipids, free cholesterol, and apolipoproteins. This composition ensures water solubility to all lipophilic components associated with HDL particles, enabling vital circulation transportation. In recent years, the list of HDL-bound components increased to proteins and enzymes, lipids, small non-coding RNAs (ncRNAs), hormones, and vitamins, making the HDL particle a very versatile but complex structure.
HDL proteomeRecent advances in proteomics and the “HDL Proteome Watch” project have increased our knowledge about HDL-bound protein species. While 85 different proteins were reported to associate with HDL particles a few years back, this number has risen to 251.28 It is important, however, to mention the impact that HDL isolation has on the reported proteomic profile. While each of the common isolation techniques (ultracentrifugation, immunoaffinity, electrophoresis, and gel filtration) has its “flaws”, cross-referencing proteomic profiles over studies and isolation techniques has enabled us to define a more robust selection of 91 “very likely” protein constituents of the HDL particle28 (Table 3).
Major components of the HDL proteome.
| Protein | UniProt ID | Found in % of studies (HDL proteome watch project)161 |
|---|---|---|
| Actin | P60709 | 31 |
| Afamin | P43652 | 41 |
| Albumin | P02768 | 84 |
| Alpha-1-antichymotrypsin | P01011 | 59 |
| Alpha-1-antitrypsin | P01009 | 94 |
| Alpha-1-microglobulin | P02760 | 75 |
| Alpha-1B-glycoprotein | P04217 | 61 |
| Alpha-2-antiplasmin | P08697 | 57 |
| Alpha-2-HS-glycoprotein | P02765 | 88 |
| Alpha-2-macroglobulin | P01023 | 61 |
| Angiotensinogen | P01019 | 69 |
| Antithrombin-III | P01008 | 55 |
| Apolipoprotein A-I | P02647 | 100 |
| Apolipoprotein A-II | P02652 | 96 |
| Apolipoprotein A-IV | P06727 | 100 |
| Apolipoprotein B-100 | P04114 | 84 |
| Apolipoprotein C-I | P02654 | 96 |
| Apolipoprotein C-II | P02655 | 92 |
| Apolipoprotein C-III | P02656 | 98 |
| Apolipoprotein C-IV | P55056 | 43 |
| Apolipoprotein D | P05090 | 82 |
| Apolipoprotein E | P02649 | 98 |
| Apolipoprotein F | Q13790 | 76 |
| Apolipoprotein L1 | O14791 | 100 |
| Apolipoprotein M | O95445 | 92 |
| Apolipoprotein(a) | P08519 | 63 |
| C4b-binding protein alpha chain | P04003 | 47 |
| Ceruloplasmin | P00450 | 39 |
| Clusterin | P10909 | 90 |
| Coagulation factor V | P12259 | 18 |
| Complement C1r subcomponent | P00736 | 29 |
| Complement C1s subcomponent | P09871 | 35 |
| Complement C3 | P01024 | 88 |
| Complement C4-A | P0C0L4 | 63 |
| Complement C4-B | P0C0L5 | 69 |
| Complement C5 | P01031 | 22 |
| Complement component C9 | P02748 | 59 |
| Complement factor B | P00751 | 41 |
| Complement factor H | P08603 | 33 |
| Complement factor I | P05156 | 22 |
| Fibrinogen alpha chain | P02671 | 78 |
| Fibrinogen beta chain | P02675 | 55 |
| Fibrinogen gamma chain | P02679 | 49 |
| Fibronectin | P02751 | 41 |
| Gelsolin | P06396 | 43 |
| Haptoglobin | P00738 | 75 |
| Haptoglobin-related protein | P00739 | 84 |
| Hemoglobin subunit alpha | P69905 | 61 |
| Hemoglobin subunit beta | P68871 | 65 |
| Hemopexin | P02790 | 65 |
| Heparin cofactor 2 | P05546 | 49 |
| Histidine-rich glycoprotein | P04196 | 39 |
| Immunoglobulin heavy constant alpha 1 | P01876 | 43 |
| Immunoglobulin heavy constant gamma 1 | P01857 | 55 |
| Immunoglobulin heavy constant gamma 2 | P01859 | 45 |
| Immunoglobulin heavy constant gamma 3 | P01860 | 29 |
| Immunoglobulin heavy constant gamma 4 | P01861 | 24 |
| Immunoglobulin heavy constant mu | P01871 | 31 |
| Immunoglobulin J chain | P01591 | 25 |
| Immunoglobulin kappa constant | P01834 | 41 |
| Immunoglobulin kappa variable 4-1 | P06312 | 16 |
| Immunoglobulin lambda constant 1 | P0CG04 | 20 |
| Immunoglobulin lambda constant 2 | P0DOY2 | 20 |
| Immunoglobulin lambda variable 3-21 | P80748 | 18 |
| Immunoglobulin lambda-like polypeptide 5 | B9A064 | 25 |
| Inter-alpha-trypsin inhibitor heavy chain H1 | P19827 | 43 |
| Inter-alpha-trypsin inhibitor heavy chain H2 | P19823 | 49 |
| Inter-alpha-trypsin inhibitor heavy chain H4 | Q14624 | 65 |
| Kallistatin | P29622 | 29 |
| Kininogen-1 | P01042 | 41 |
| Lipopolysaccharide-binding protein | P18428 | 37 |
| Phosphatidylinositol-glycan-specific phospholipase D | P80108 | 51 |
| Phospholipid transfer protein | P55058 | 71 |
| Plasma kallikrein | P03952 | 33 |
| Plasma protease C1 inhibitor | P05155 | 55 |
| Plasminogen | P00747 | 41 |
| Pregnancy zone protein | P20742 | 24 |
| Prothrombin | P00734 | 63 |
| Retinol-binding protein 4 | P02753 | 61 |
| Serotransferrin | P02787 | 84 |
| Serum amyloid A-1 protein | P0DJI8 | 84 |
| Serum amyloid A-2 protein | P0DJI9 | 84 |
| Serum amyloid A-4 protein | P35542 | 88 |
| Serum amyloid P-component | P02743 | 24 |
| Serum paraoxonase/arylesterase 1 | P27169 | 94 |
| Serum paraoxonase/lactonase 3 | Q15166 | 59 |
| Transthyretin | P02766 | 82 |
| Vitamin D-binding protein | P02774 | 78 |
| Vitamin K-dependent protein S | P07225 | 24 |
| Vitronectin | P04004 | 73 |
| Zinc-alpha-2-glycoprotein | P25311 | 35 |
91 most reproducible HDL-associated proteins identified with all four standard isolation methods including electrophoresis, filtration, immunoaffinity, and ultracentrifugation. Data represented in alphabetical order.
Recent and extensive work suggests HDL subclasses according to the main functions associated with respective protein clusters.28 The biggest and most important subclasses include proteins involved in (1) lipid transportation (e.g., ApoA-I/-II, ApoC-I/-II/-III, ApoE, ApoM, CETP, LCAT) being ApoA-I and ApoA-II the most important structural pillars in all HDL particles; (2) homeostasis/protease inhibition (e.g., ITIH1/2/3, coagulation factors such as F5 and F13B); (3) inflammation/acute phase response (e.g., serum amyloid A1/2/4, ITIH4); (4) immunity/anti-microbial (e.g., immunoglobulins such as IGKV3-20, IGHV3-13, and complement pathway components such as C3, C4B, C6); and (5) cell/heparin-binding (e.g., SELL, DSG1, CD44, ANGPTL3). However, functions that are less represented, such as vitamin binding and transport, including the vitamin D- and retinol-binding proteins, or metal ion binding, need also to be considered. Nevertheless, whether strongly or weakly represented, the countless functions highlight important learning from the last years of research: HDL particles are much more than just lipid transporters.
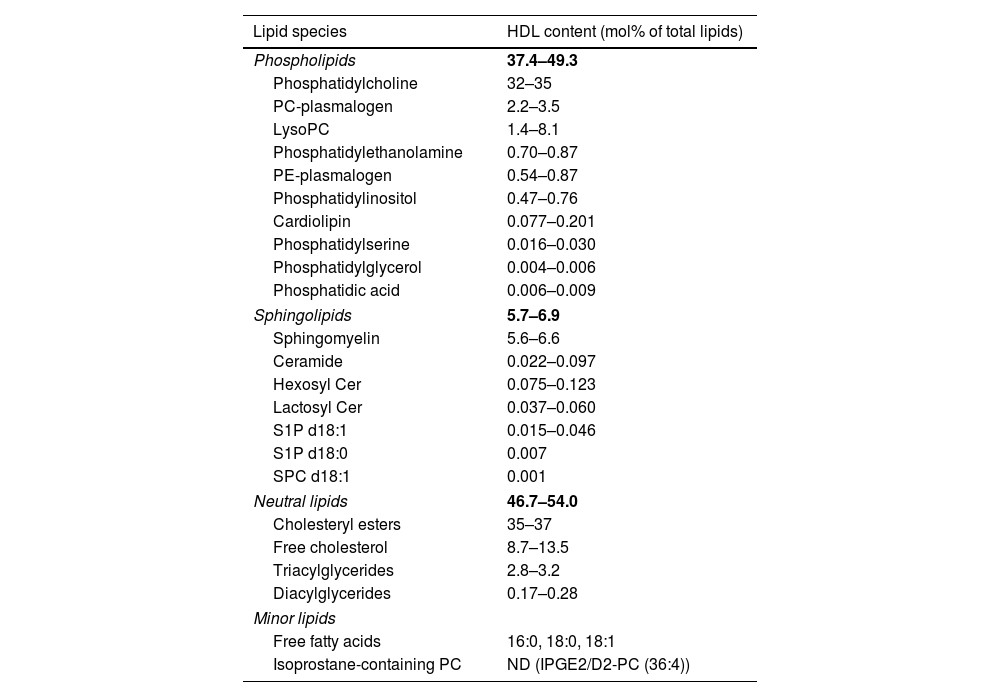
HDL lipidomeThe HDL lipidome is just as diverse as its proteomic counterpart, consisting of more than 400 HDL-bound lipid species identified so far.29 Based on their chemical properties, amphiphilic lipids such as free cholesterol and phospholipids are carried on the surface of the HDL particle, protecting the neutral hydrophobic lipids such as cholesteryl esters and triglycerides in the HDL core. The major HDL-bound lipid species are phospholipids with 37.4–49.3mol%, cholesteryl esters with 35.0–37.0mol%, free cholesterol with 8.7–13.5mol%, sphingolipids with 5.7–6.9mol%, and triglycerides with 2.8–3.2mol% of total lipids.30 Phospholipids and sphingolipids form a big part of the HDL lipidome, with phosphatidylcholine and sphingomyelin being the most abundant species, respectively.30 The HDL lipidome constitutes many more lipids, though to a much lower abundance (Table 4).
Major components of the HDL lipidome.
| Lipid species | HDL content (mol% of total lipids) |
|---|---|
| Phospholipids | 37.4–49.3 |
| Phosphatidylcholine | 32–35 |
| PC-plasmalogen | 2.2–3.5 |
| LysoPC | 1.4–8.1 |
| Phosphatidylethanolamine | 0.70–0.87 |
| PE-plasmalogen | 0.54–0.87 |
| Phosphatidylinositol | 0.47–0.76 |
| Cardiolipin | 0.077–0.201 |
| Phosphatidylserine | 0.016–0.030 |
| Phosphatidylglycerol | 0.004–0.006 |
| Phosphatidic acid | 0.006–0.009 |
| Sphingolipids | 5.7–6.9 |
| Sphingomyelin | 5.6–6.6 |
| Ceramide | 0.022–0.097 |
| Hexosyl Cer | 0.075–0.123 |
| Lactosyl Cer | 0.037–0.060 |
| S1P d18:1 | 0.015–0.046 |
| S1P d18:0 | 0.007 |
| SPC d18:1 | 0.001 |
| Neutral lipids | 46.7–54.0 |
| Cholesteryl esters | 35–37 |
| Free cholesterol | 8.7–13.5 |
| Triacylglycerides | 2.8–3.2 |
| Diacylglycerides | 0.17–0.28 |
| Minor lipids | |
| Free fatty acids | 16:0, 18:0, 18:1 |
| Isoprostane-containing PC | ND (IPGE2/D2-PC (36:4)) |
The lipids are ordered according to lipid class and abundance. PC: phosphatidylcholine; PE: phosphatidylethanolamine; S1P: sphingosine-1-phosphate; SPC: sphingosylphosphorylcholine.
Sphingomyelin and free cholesterol, two lipidic components that affect membrane fluidity, decrease with increasing density of HDL particles (HDL2→HDL3).31 Hence, denser HDL3 particles exert better membrane fluidity, which may have functional implications for interactions with receptors and transporters.31 Likewise, the abundance of sphingosine-1-phosphate (S1P) per HDL particle is significantly higher in dense HDL3 (40–50mmol/mol HDL) particles compared with HDL2 particles (15–20mmol/mol).31–33 The enrichment of small, dense HDL3 with S1P is driven by HDL-bound ApoM, one of the main S1P carriers in circulation,34 which is predominantly associated with dense HDL3 particles35 and has functional implications.36
HDL-C measured in routine clinical analysis reflects the amount of cholesterol levels transported HDL particles but not whether they are functional.37 As discussed below, the assessment of HDL function would better reflect HDL's capability to exert cardiovascular protective effects.
Other HDL constituentsAlthough HDL particles are primarily known for their lipid-transporting properties, they carry and deliver many other components, including hormones, carotenoids, vitamins, and ncRNAs.38 The very first hormone to be identified on HDL particles was the thyroid hormone thyroxine (T4), which was shown to interact with apolipoprotein moieties (ApoA, ApoC) and to reduce binding upon increasing lipid content.39,40 LCAT-mediated esterification of the hormone (derivates) oestrogen, pregnenolone, and dehydroepiandrosterone (fatty acid ester versions) have also been detected on HDL particles.41–43 The physiological functions of these HDL-bound hormones remain largely unknown; they might, however, serve as precursors for cellular steroid synthesis or transcriptional regulators acting on nuclear receptor transcription factors once they have been delivered to the target cell by a potentially SR-B1-dependent mechanism.44
Furthermore, HDL has very recently been shown to be the primary transporter for carotenoids (β-carotene, zeaxanthin, and lutein in decreasing binding affinity to ApoA-I) from the liver to the retinal pigment epithelium where they protect against macular degeneration and blindness.45 In addition to these antioxidants, α-carotene, lycopene, and cryptoxanthin can also be transported by HDL particles.46 HDL particles transport more polar carotenoids (53% lutein, 39% cryptoxanthin) than nonpolar carotenoids (17% lycopene, 26% α-carotene, 22% β-carotene).44
Carotenes and cryptoxanthin are provitamin A carotenoids that can be converted into fat-soluble vitamin A, essential in conferring growth, vision, cell division, immunity, and reproduction. In addition, carotenes have also been demonstrated to decrease atherosclerotic risk in humans,47 delay atherosclerosis progression,48 and increase atherosclerosis resolution49 in mice.
Nevertheless, vitamin A is not the only member of the fat-soluble vitamins that is transported in HDLs. Vitamin E (α-tocopherol) is probably the most important HDL-transported lipid-soluble antioxidant protecting cells and susceptible molecules from free radicals.50,51 Mechanistically, vitamin E might interact with HDL-bound ApoA-I, as its transport is inhibited by ABC transporter inhibition.52 However, while vitamin E delivery to epithelial cells has been reported to be SR-B1-independent,53 delivery to endothelial cells was shown to be dependent on SR-B1.54,55
On the other hand, vitamin D is not a typical HDL constituent since the serum protein vitamin D-binding protein mainly carries this vitamin. However, its carrier has been identified as part of the HDL proteome56,57 and can, therefore, be considered part of the HDL particle.
In recent years, HDL particles have also been shown to carry many types of small ncRNAs, amongst which microRNAs (miRNAs) are the second most abundant ncRNA class on HDL particles after ribosomal RNA-derived ncRNA. The combination and abundance of HDL-transported miRNAs is not just a representation of the donor cells58 but that they differ from cellular miRNAs in their post-transcriptional modification (non-templated 3′-uridylation versus -adenylation, respectively).59 Although it is difficult to identify the cellular origin of HDL-transported miRNAs, the top-most abundant miRNAs on human HDL particles60 have a restricted cell type-specific expression profile61: beta cells (miR-375-3p),60 macrophages and neutrophils (miR-223-3p),60,62 and neurons (miR-124-3p, miR-9-5p).63 So far, endothelial cells,64–66 hepatocytes,60 and microglia63 are cell types with confirmed uptake of HDL-transported miRNAs and subsequent downstream target inhibition. The mechanisms involved in miRNA delivery remain unclear, although SR-B1 has been suggested to play a key role in miRNA-HDL loading.60,64 As such, global SR-B1 deficiency has been shown to be associated with a lack of accumulation of miRNAs in circulating HDL particles.59,60 Besides intensive research to identify the cross-talk between HDL-related miRNAs and cells efforts are being made to establish HDL-transported miRNAs as biomarkers for CVD.67
Cardiovascular protective functions of the HDL particleSince the ground-breaking Framingham Heart Study in the 1960–1980s,68 several large-scale observational and experimental studies have accredited HDL particles with beneficial properties in the CV system. Although the hallmark protective of HDL particles is their ability to efflux cholesterol and mediate reverse cholesterol transport (RCT),69,70 they possess further athero- and cardio-protective functions, including protection against oxidative and inflammatory damage,71–74 inhibition of thrombosis,75,76 cardioprotection,77 as well as induction of NO synthesis72,78 and endothelial cell renewal.72,79
Reverse cholesterol transportHDL particles are the key molecules in reverse cholesterol transport, as they function as cholesterol acceptors, carriers for circulation, and deliverers to the liver for biliary excretion. As such, HDL particles facilitate cholesterol efflux from peripheral tissues, including atherosclerotic foam cells, in various ways. Upon normal cholesterol levels, passive aqueous diffusion of free cholesterol between membranes and HDL particles occurs following the concentration gradient.80 Free cholesterol is translocated into the particle core by LCAT-dependent transformation into cholesteryl esters to maintain the efflux from the cells to the HDL particle. In addition, SR-B1-dependent non-aqueous efflux may occur, a pathway promoted by low total HDL levels and large HDL particles.81 In conditions with elevated cellular cholesterol concentrations (e.g., foam cells), active cholesterol efflux pathways dependent on the ABC transporter family members A1 and G1 are activated.80 Further LCAT activity leads to particle maturation and cholesteryl ester enrichment in the lipid core.
Antioxidant and anti-inflammatory capacityOxidative stress (ROS, oxLDL) and a pro-inflammatory environment (cytokines, chemokines, and endothelial adhesion molecules) drive atherosclerosis initiation and progression. HDL particles provide potent atheroprotection and prevent LDLs lipid oxidation (HDL3>HDL2) by transferring those oxidised lipids to the HDL particle, where redox-active ApoA-I residues can inactivate them.71,82 In addition, HDL particles carry multiple antioxidant enzymes such as serum paraoxonase 1 (PON1),83 lipoprotein-associated phospholipid A2,84 and LCAT.85 PON1 is the most investigated HDL-associated enzyme and equally protects HDL and LDL particles by hydrolysing lactones, phosphate esters, and lipid peroxide derivates.86 The presence or interaction with ApoA-I seems crucial since particles containing ApoA-II or -IV have shown significantly less antioxidative protection.86,87
Interestingly, PON1 has also been shown to play a role in the classical activation of eNOS through the ApoA-I/SR-B1 axis.86 Moreover, HDL-associated phospholipids may also be involved in eNOS activation by S1P3 signalling.88 Both pathways lead to HDL-mediated eNOS stimulation by converging the non-receptor tyrosine kinase Src-activated Akt and MAP kinases.89
HDL particles also have a crucial role in inhibiting the expression of the inflammatory adhesion molecules VCAM-1 and ICAM-1,90 E-selectin,91 and MCP-192 on activated endothelial cells thereby preventing the recruiting and extravasation of immune cells into the intimal space. The suggested molecular mechanisms behind are HDL-S1P-dependent and involve the interference with NFκB signalling (S1P1/β-arrestin2 axis93) and a reduction in endothelial apoptosis and TNF-mediated inflammatory signalling (P13K/AKT/eNOS axis94,95). HDL-S1P has also been reported to inhibit macrophage apoptosis through JAK2/STAT3 signalling.96 Whereas HDL-related inhibition of endothelial inflammatory surface molecules seems independent of variations in HDL size and composition of apolipoproteins, cholesteryl ester, and triglycerides,97 phospholipid subspecies have shown to exert antiinflammatory effects98 when protected from oxidation by either ApoA-I or other antioxidants.99
Antithrombotic effectsHDL particles exert several antithrombotic effects that may protect against atherothrombotic clinical events. As such, HDL particles promote blood flow by increased NO and prostacyclin synthesis with subsequent vasodilation. HDL also prevent endothelial cell activation by inhibiting endothelial cell apoptosis89 and the expression of the prothrombotic factors P-selectin,100 E-selectin,100 and tissue factor.101 NO and prostacyclin also inhibit platelet activation and aggregation,102 which, together with their vasodilator properties, counterbalance restricted blood flow. In addition, HDL particles have been reported to upregulate endothelial thrombomodulin,103 an anticoagulant factor that inhibits thrombin formation.
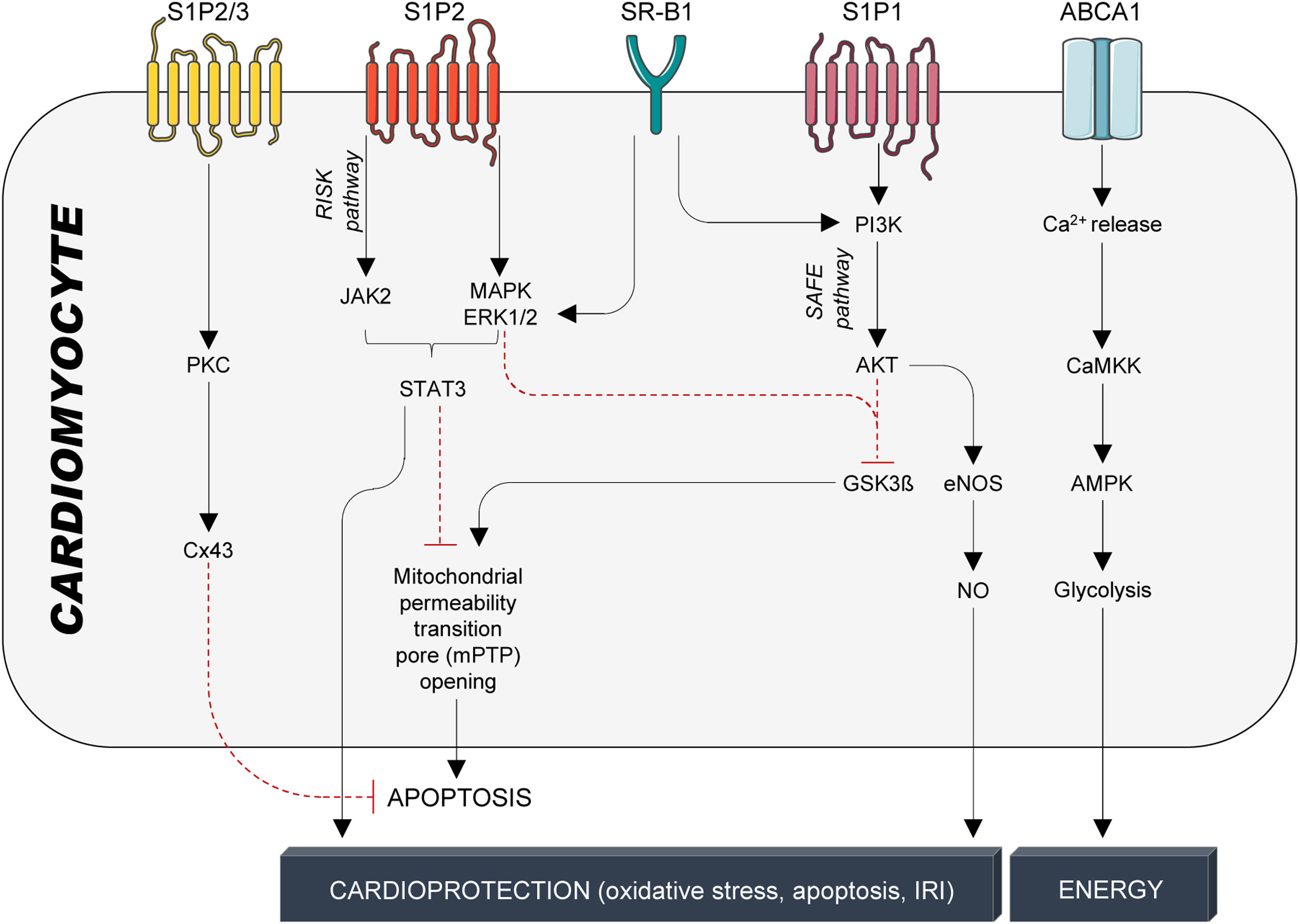
Cardioprotection: protection against ischemia–reperfusion injuryWhile cardiac tissue and performance are more imminently threatened during ischemia,104 reperfusion – although indispensable for saving ischemic heart tissue from cell death – further damages the injured heart tissue.105 Consequently, the total damage of ischemia–reperfusion injury (IRI) is the sum of the ischemic insult (restricted or interrupted blood flow) and reperfusion (restored blood flow and re-oxygenation).106 Early experimental studies in rodents reported that intravenous infusions of HDL particles strongly reduced infarct size77,107 and improved post-ischemic cardiac function.108 The mechanisms behind involve the reduction in apoptosis execution through activation of the reperfusion injury signalling kinase (RISK) and survivor activating factor enhancement (SAFE) signalling pathways109 (Fig. 4). The RISK pathway (P13K/AKT/eNOS axis) protects endothelial cells from pro-inflammatory TNFα signalling, whereas the SAFE pathway is likely to be activated by HDL-bound S1P110 with TNFα and STAT3 downstream signalling effectors.111 STAT3 activation is involved in the early protection phase by preventing the opening of the mitochondrial permeability transition pore and subsequent apoptosis execution, and in the late protective phase, in which STAT3 phosphorylation leads to expression of target genes, eventually reducing oxidative stress, apoptosis, and IRI110 (Fig. 4). Furthermore, HDL-S1P signalling also prevents cardiomyocyte cell death and significantly reduces infarct size by phosphorylating connexin 43 (Cx43), the main protein of cardiomyocyte Cx43 gap junction channels112 (Fig. 4).

HDL-mediated protective signalling in cardiomyocytes. Protective signalling in cardiomyocytes is mediated through the G-protein coupled receptors S1P1-3 and the cholesterol transporters SR-B1 and ABCA1. S1P1-3 are activated by the interaction with S1P, which is bound to HDL particles by its interaction with ApoM. SR-B1 and ABCA1, on the other hand, are activated by binding to ApoA-I, the main structural protein of HDL particles and the main inducer of active cholesterol efflux. HDL particles confer cardioprotection in cardiomyocytes through a multitude of kinase cascades that reduce stressors, prevent cardiomyocyte apoptosis, and, therefore, improve infarct size, cardiac function, and overall outcome. S1P1-3: Sphingosine-1-phosphate receptors 1-3; SR-B1: scavenger receptor B1; ABCA1: ATP-binding cassette transporter A1; PKC: protein kinase C; Cx43: connexin 43; JAK2; Janus kinase 2; MAPK: mitogen-activated protein kinase; ERK1/2: extracellular signal-regulated kinase 1/2; STAT3: signal transducer and activator of transcription 3; PI3K: phosphatidylinositol-4,5-bisphosphate 3-kinase; AKT: serine/threonine kinase 1; GSK3β: inosine/guanosine kinase 3β; eNOS: endothelial nitric oxide synthase; NO: nitric oxide; CaMKK: calcium/calmodulin-activated protein kinase; AMPK: adenosine monophosphate-activated protein kinase.
Evidence also suggests that HDL-transported miRNAs are involved in protection against IRI. As such, inhibition of miR-92a, a miRNA reported to be carried by HDL particles, has been shown in small and large animal models to reduce infarct size and improve post-ischemic cardiac function.113–116 Likewise, miR-486 has also been shown to attenuate IRI by reducing cardiomyocyte apoptosis in vitro117,118 and in vivo118 and also mediate beneficial effects on exercise-induced myocardial protection.118 Furthermore, recent advances also suggest protective effects for the presence of miR-146a119,120 and miR-125,121,122 both part of an HDL-miRNA profile identified in coronary artery disease patients.67
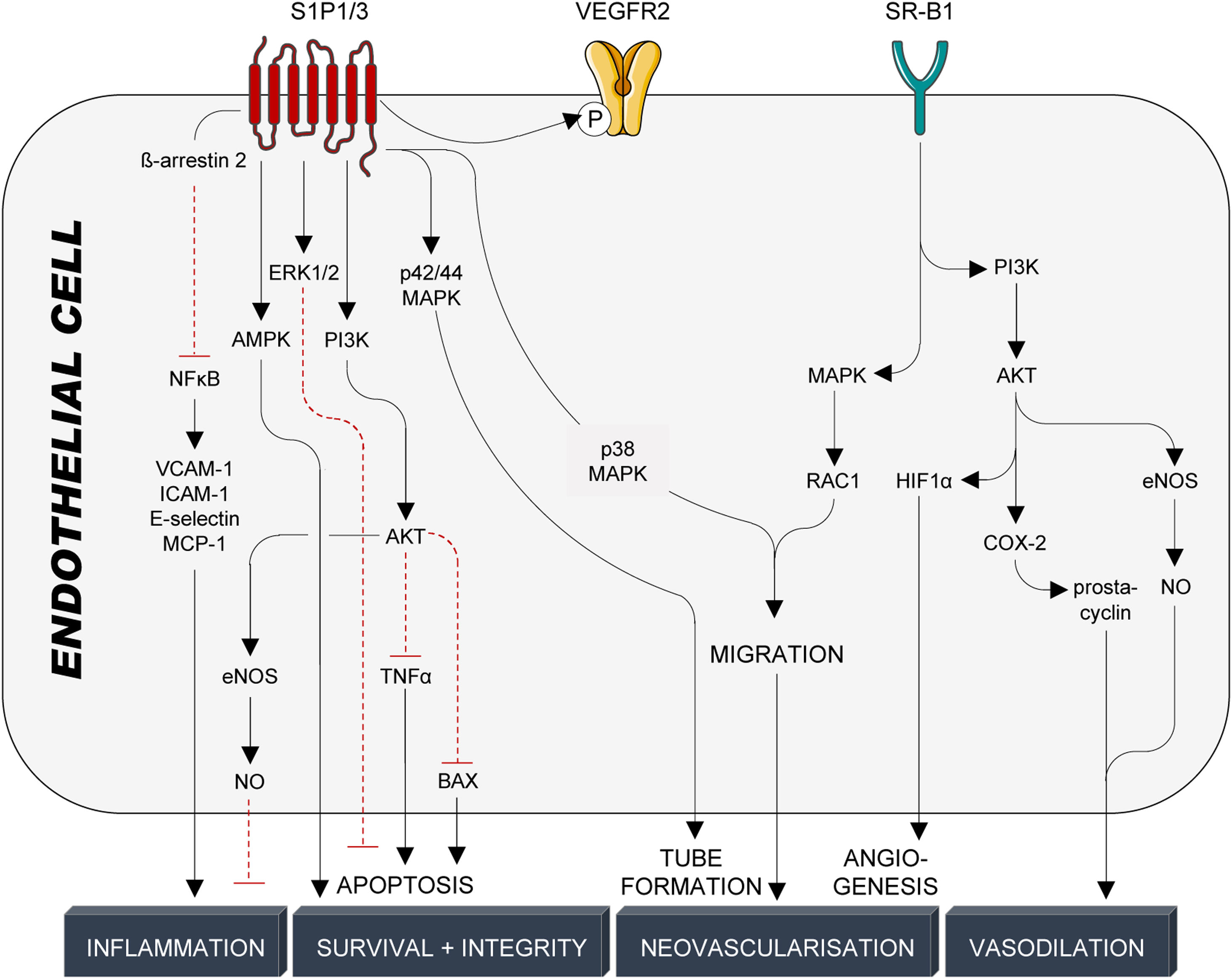
Endothelial protectionThe endothelium forms a semipermeable barrier that separates blood-carried factors and cells from the surrounding tissue123 and its integrity is essential for sustaining vascular homeostasis, including the modulation of vascular tone and trafficking of substances between systemic circulation and adjacent tissues.124 HDL particles support endothelial monolayer integrity by promoting junction closure,125 preventing loss of glycocalyx (proteoglycan shedding),126 reducing endothelial cell apoptosis,110 and stimulating endothelial cell proliferation and migration of both mature endothelial cells127 as well as endothelial progenitor cells.128 As part of their antiapoptotic properties, HDL particles prevent sustained increases of intracellular calcium,89 the activation of caspases 3 and 9129,130 (TNFα- and growth factor deprivation-mediated apoptosis), and modulate S1P-dependent activation of PI3K/AKT94,110,131 and ERK1/2.132 On the other hand, endothelial cell proliferation and migration are crucial for neovascularisation and reendothelisation after vascular injury.127 HDL stimulation of endothelial cell migration and proliferation is mediated by multiple kinase cascades involving SR-B1-dependent activation of MAPK/Rac1133 (migration and re-endothelialisation), PI3K/AKT134 (HIF1α-mediated angiogenesis), as well as S1P1/3-dependent activation of ERK135 (endothelial cell survival), PI3K/AKT,135 p38 MAPK,135 Rho/Rho kinase135 (endothelial cell migration), p42/44 MAPK/Ras136 (tube formation), AKT125 and AMPK137 (endothelial barrier integrity), NFκB90,95 and PI3K/AKT/eNOS110 (anti-inflammatory, decrease in surface expression of VCAM-1, ICAM-1, E-selectin, MCP-1), and VEGFR2138 (angiogenesis) (Fig. 5). As interference with the S1P/S1P1/3 axis mimics effects seen by interference with SR-B1, HDL particles were suggested to primarily interact with the SR-B1 receptor on endothelial cells, which secondarily leads to spatial proximity between HDL-bound S1P and endothelial S1P1/3 receptors. As stated above, endothelial cells are important SR-B1-dependent inducers of NO and prostacyclin synthesis, which strongly contribute to PDZK1/SRC/AKT/MAPK/eNOS-139 and PDZK1/AMPK/PI3K/AKT/COX-2/prostacyclin-140 as well as SphK2/COX-2/prostacyclin141-mediated vasodilation (Fig. 5).

HDL-mediated protective signalling in endothelial cells. Protective signalling in endothelial cells is mediated through the G-protein coupled receptors S1P1/3 and the cholesterol transporter SR-B1. S1P1/3 are activated by the interaction with S1P, which is bound to HDL particles by its interaction with ApoM. SR-B1, on the other hand, is activated by binding to ApoA-I, the main structural protein of HDL particles and the main inducer of active cholesterol efflux. HDL particles confer cardioprotection in endothelial cells through a multitude of kinase cascades, NFκB and VEGFR2, that prevent endothelial cell activation and inflammation, promote endothelial cell survival and barrier integrity and induce tube formation, migration, and angiogenesis. Production and release of NO and prostacyclin from endothelial cells also trigger vascular smooth muscle cell relaxation and subsequent vasodilation. S1P1/3: sphingosine-1-phosphate receptors 1/3; VEGFR2: vascular endothelial growth factor receptor 2; SR-B1: scavenger receptor B1; NFκB: nuclear factor κB; VCAM-1: vascular cell adhesion molecule 1; ICAM-1: intracellular cell adhesion molecule 1; MCP-1: monocyte chemoattractant protein 1; AMPK: adenosine monophosphate-activated protein kinase; ERK1/2: extracellular signal-regulated kinase 1/2; PI3K: phosphatidylinositol-4,5-bisphosphate 3-kinase; AKT: serine/threonine kinase 1; TNFα: tumor necrosis factor α; BAX: Bcl-2-associated X protein; eNOS: endothelial nitric oxide synthase; NO: nitric oxide; MAPK: mitogen-activated protein kinase; RAC1: Ras-related C3 botulinum toxin substrate 1; HIF1α: hypoxia-inducible factor 1α; COX-2: cyclooxygenase-2.
The concept that raising HDL-C would promote protection against CVD was developed from observing several large epidemiological studies that evidenced that low HDL-C levels strongly correlated with future CV events in healthy individuals without baseline CVD.68,142–144 However, doubts arose when extremely high HDL-C levels were paradoxically shown to increase all-cause and CV mortality in both men and women145 and Mendelian randomisation studies failed to confirm a causative interaction between increased CVD risk and HDL-C.146 Furthermore, extensive efforts to pharmacologically raise HDL-C levels through several randomized clinical trials (i.e., niacin, CETP inhibitors) failed to reduce CV events in secondary prevention.147–150
The failure of the cetrapib trialsThe efficacy of several CETP inhibitors (torcetrapib, dalcetrapib, evacetrapib, and anacetrapib) was tested in four large-scale phase-III clinical trials148–151 on top of statin treatment. The hypothesis was that while statins would limit atherosclerotic disease progression, HDL-C-raising treatment would simultaneously work on reversing the existing atherosclerotic plaques. Besides, CETP inhibitors would also minimise the residual CV risk in high-risk patients on statins with successfully lowered LDL-C levels but low HDL-C levels.
The first trial was the ILLUMINATE (Investigation of Lipid Level Management to Understand its Impact in Atherosclerotic Events) trial, which administered torcetrapib in combination with atorvastatin to 15,067 patients for 12 months.150 Despite significantly increased HDL-C (70%) and decreased LDL-C (25%), torcetrapib treatment elevated the risk for major adverse CV events (MACE) and CVD-related as well as unrelated deaths compared to the placebo group.150 As a result, the trial was terminated prematurely.
In the dal-OUTCOME (Study of RO4607381 in Stable Coronary Heart Disease Patients With Recent Acute Coronary Syndrome) trial, dalcetrapib or placebo was administered to patients on statins and with a recent acute coronary syndrome for a mean of 31 months.149 Despite a significant increase in HDL-C levels by 31–40% and fewer adverse effects than torcetrapib, the trial was terminated due to futility as no significant reduction in hard primary endpoints was detected. Nevertheless, later studies found that a minority of 17% of the cohort significantly benefited from dalcetrapib treatment (39% reduction in CV endpoints) due to a homozygous single nucleotide morphism in the adenylate cyclase nine gene.152
The ACCELERATE (Assessment of Clinical Effects of Cholesteryl Ester Transfer Protein Inhibition with evacetrapib in Patients at a High Risk for Vascular Outcomes) trial administered anacetrapib or placebo to 12,092 patients on statins and with high-risk vascular disease (over 63% of the patients presented with diabetes) for a mean period of 26 months.148 Despite the massive change in lipoprotein cholesterol levels (HDL-C +133%; LDL-C −31%) and significantly lower incidence rates of death from any cause in anacetrapib-treated patients, the ACCELERATE trial did not lead to reduced primary CV endpoints. It was terminated after two years of follow-up due to a lack of efficacy.
Finally, the REVEAL (Randomized Evaluation of the Effects of Anacetrapib through Lipid-modification) trial was the longest and largest trial, with 30,449 patients on atorvastatin and with established atherosclerotic CVD receiving anacetrapib or placebo for a median period of over four years.151 Anacetrapib strongly modified the lipid profile (HDL-C +104%, LDL-C −41%) and was the first to report modest but significantly reduced primary endpoint rates, including coronary death, myocardial infarction, and coronary revascularisation in anacetrapib-treated patients (10.8% vs 11.8% in placebo, p=0.004). However, the benefit was small and only significant when combining data from all years or years>1, which questioned the usefulness for high-risk patients needing short-term solutions. In the end, the reduction in the primary CV endpoint was attributed to the drop in LDL-C levels rather than the rise of HDL-C.
The results of the trials above failed to meet the expectations for therapeutically raising HDL-C levels in high-risk patients on statin treatment. Furthermore, a meta-analysis of over 113,000 statin-treated patients in 39 trials involving the HDL-C-raising therapies niacin, fibrates, and CETP inhibitors demonstrated no difference in CV events between those patients on these HDL-C-related therapies and placebo controls.147 The beneficial effect that was provided by lowering LDL-C (by statin or otherwise) was strongly diminished by very high HDL-C (in putatively dysfunctional HDL), as seen in the REVEAL trial and suggested for both the ACCELERATE and dal-OUTCOME trials. It is essential to consider patients’ additional CV risk factors and comorbidities on top of dyslipidaemia. In this regard, besides hypertension and chronic kidney disease, two out of four CETP inhibitors promoted de novo onset of diabetes, likely increasing the patient's risk.
In summary, cetrapibs were developed assuming that increasing HDL-C would translate into enhanced hepatic cholesterol excretion and subsequent cardio/vascular protection. CETP inhibitors block cholesteryl ester transport from HDL to VLDL, IDL, and LDL particles to enhance the amount of cholesterol reaching the liver (Fig. 6). However, in turn, CETP inhibition leads to the formation of large cholesteryl ester-enriched particles that, in contrast to the smaller and denser HDL subfractions (i.e., HDL3), have been shown to not interact efficiently with ABCA1, the major mediator of cholesterol efflux from macrophages to HDLs153 (Fig. 6).
. The cholesteryl ester transfer protein (CETP) is an enzyme that catalyzes the transport of cholesteryl esters and triglycerides between lipoproteins. Delivery of HDL-transported cholesteryl ester to ApoB-containing particles (LDLs/IDLs/VLDLs) is mediated by CETP in exchange for triglycerides. This process is selectively blocked by the drug family of CETP inhibitors, also known as cetrapibs, and leads to the formation of large, cholesteryl ester-enriched HDL particles. These particles lack efficient interaction with the ABC transporter family, the major mediators of cholesterol efflux from macrophages to HDL particles, subsequently reducing (cardio-) vascular protection via reverse cholesterol transport. ABCG1: ATP-binding cassette transporter G1; SR-B1: scavenger receptor B1; LCAT: lecithin cholesteryl acetyltransferase; CE: cholesteryl ester; HDL: high-density lipoprotein; LDL: low-density lipoprotein; IDL: intermediate-density lipoprotein; VLDL: very low-density lipoprotein; LDL-R: LDL receptor.")
Mechanism of CETP inhibitors (cetrapibs). The cholesteryl ester transfer protein (CETP) is an enzyme that catalyzes the transport of cholesteryl esters and triglycerides between lipoproteins. Delivery of HDL-transported cholesteryl ester to ApoB-containing particles (LDLs/IDLs/VLDLs) is mediated by CETP in exchange for triglycerides. This process is selectively blocked by the drug family of CETP inhibitors, also known as cetrapibs, and leads to the formation of large, cholesteryl ester-enriched HDL particles. These particles lack efficient interaction with the ABC transporter family, the major mediators of cholesterol efflux from macrophages to HDL particles, subsequently reducing (cardio-) vascular protection via reverse cholesterol transport. ABCG1: ATP-binding cassette transporter G1; SR-B1: scavenger receptor B1; LCAT: lecithin cholesteryl acetyltransferase; CE: cholesteryl ester; HDL: high-density lipoprotein; LDL: low-density lipoprotein; IDL: intermediate-density lipoprotein; VLDL: very low-density lipoprotein; LDL-R: LDL receptor.
Pioneering studies from our group demonstrated that administration of cholesterol-poor HDL particles (equivalent to HDL3 particles) isolated from healthy animals limited further lipid deposition and even regressed atherosclerotic lesions.70,154 These and other experimental studies have supported the “HDL quality over quantity hypothesis”. The scientific field has accordingly redirected its efforts towards deciphering HDL structure and function to restore HDL-related CV protection,155 particularly since multiple studies have suggested that pathophysiologic conditions adversely remodel HDL particles [reviewed in30,82,156], thus impairing HDL protective effects.82,156 In this regard, we have demonstrated in a highly translatable pig model that diet-induced hypercholesterolemia, one of the most prevalent CV risk factors, render HDL particles dysfunctional and induce compositional changes at a proteomic, lipidomic and miRNA level.108 Interestingly, we have also evidenced that such adverse HDL remodelling can be reversed and cardiovascular protection restored by feeding a low-fat diet and decreasing LDL-C levels back to the physiological state.157
ConclusionsThe landmark Framingham Heart Study evidenced an inverse association between CV risk and HDL-C plasma levels, fostering experimental research towards deciphering the beneficial properties of HDL particles. However, excitement towards the therapeutic potential of HDL-C-raising drugs faded with the disappointing outcomes of the cetrapib trials. These data evidenced no clear benefit of increasing HDL-C in patients with established CVD in secondary prevention and, most importantly, questioned whether HDL-C was the adequate parameter to reflect HDL-related protection.158 HDL is a complex and heterogeneous particle with many components that exert various cellular and molecular functions beyond cholesterol transport and removal.82,156 The presence of CV risk factors and comorbidities have been shown to remodel HDL particles towards a dysfunctional state, yet, experimental evidence supports the ability to restore HDL protective function by implementing healthy habits re-opening the door for new considerations in CVD prevention.157 Further research is warranted to identify how these particles can rise to their former glory and maintain their protective phenotype in primary and secondary prevention.
FundingThis work was supported by the Spanish Society of Atherosclerosis/Atherosclerosis Spanish Foundation (Research Grant SEA/FEA 2019), and PID2021-128891OB-I00 (to GV), PID2019-107160RB-I00 (to LB), and PLEC2021-007664 NextGenerationEU (to GV) funded by MCIN/AEI/10.13039/501100011033 and Fondo Europeo de Desarrollo Regional (FEDER) A way of making Europe; the Instituto de Salud Carlos III [CIBERCV CB16/11/00411 to LB]; the Generalitat of Catalunya-Secretaria d’Universitats i Recerca del Departament d’Economia i Coneixement de la Generalitat [2017SGR1480 to LB] and 2016PROD00043 (Agencia Gestión Ayudas Universitarias Investigación: AGAUR), CERCA programme/Generalitat de Cataluña, and Fundación Investigación Cardiovascular - Fundación Jesús Serra for their continuous support.
Conflict of interestThe authors have no conflict of interest to declare related to this manuscript.