


Recientemente, la tecnología conocida como array-CGH se ha establecido como una herramienta diagnóstica de primer orden para el estudio de pacientes con anomalías congénitas, retraso mental no filiado y otras enfermedades neurológicas. Sin embargo, su utilidad como técnica de primer uso en el campo prenatal está actualmente en fase de evaluación, especialmente en embarazos de bajo riesgo. En una población de 530 gestantes con embarazos de bajo riesgo se realizó, simultáneamente, cariotipo convencional y un estudio de array-CGH para el diagnóstico prenatal. Mientras que el cariotipo detectó 3 casos (0,5%) con alteraciones citogéneticas no equilibradas (una de ellas no definida), el array-CGH detectó 8 casos con este tipo de alteraciones (1,5%), identificando el cambio indefinido detectado por cariotipo. Este estudio demuestra positivamente que el array-CGH puede ser una herramienta útil en el diagnóstico prenatal en embarazos de bajo riesgo.
The array-CGH technique has recently been established as a first-tier diagnostic test for studying patients with congenital anomalies, idiopathic mental retardation and other neurological disorders. However, its use in prenatal diagnosis is still being evaluated, especially in low-risk pregnancies. A study was conducted on a population of 530 low-risk pregnancy women using both conventional karyotype and array-CGH for prenatal diagnosis. Whereas conventional karyotype detected 3 foetuses (0.5%) with unbalanced cytogenetic aberrations (one of them was undefined), array-CGH detected 8 foetuses with copy number aberrations (1.5%), and positively identified the undefined cytogenetic aberration detected using karyotype. In conclusion, this study proposes array-CGH as a useful tool in prenatal diagnosis for low-risk pregnancies.
El diagnóstico genético es una herramienta que permite conocer la presencia de alteraciones genéticas en un individuo afecto de una enfermedad, presumiblemente genética, y asociarlas a un fenotipo concreto, así como a una predicción de la evolución clínica y del pronóstico del individuo1. Dentro del área del diagnóstico genético, se encuentra el estudio prenatal de anomalías cromosómicas, que se encarga de evaluar las cromosomopatías presentes en el ADN fetal, para evaluar la posibilidad de que un feto, antes de su nacimiento, presente un síndrome malformativo determinado, asociado a un espectro fenotípico y evolución concretos2.
Actualmente, el estudio de anomalías cromosómicas a nivel prenatal precisa de una técnica invasiva para obtener material genético fetal, ya sea a través de células fetales descamadas en el líquido amniótico3, a través de una biopsia de las vellosidades coriónicas4, o a través de una extracción de sangre del cordón umbilical o cordocentesis5. Estas técnicas tienen un riesgo, tanto para el feto como para la gestante, que, aunque reducido, debe ser tenido en cuenta a la hora de evaluar si es necesaria dicha técnica invasiva6. Por ello, previamente al diagnóstico de anomalías cromosómicas, existen una serie de tests de cribado, basados en parámetros de edad materna, bioquímica y ecografía, para evaluar el riesgo para el feto de presentar un síndrome malformativo determinado, como el síndrome de Down o el síndrome de Edwards, identificable a nivel cromosómico, que justifique la técnica invasiva7.
Las técnicas de detección de anomalías cromosómicas más utilizadas actualmente son el cariotipo convencional, o cariotipo de bandeo G, y el estudio de los cinco cromosomas involucrados en las aneuploidías más frecuentes (cromosomas 13, 18, 21, X e Y) mediante hibridación in situ fluorescente (FISH), o mediante PCR fluorescente cuantitativa (QF-PCR). El cariotipo convencional permite, mediante un cultivo celular del material fetal, la obtención de metafases para la detección de cromosomopatías, con un límite de resolución bajo (más de 5 megabases)8; las otras 2 técnicas de análisis de aneuploidías cuentan con la ventaja de no precisar un cultivo celular, lo que incrementa la velocidad del análisis (dando un resultado en 48h), pero solo analizan el estado de número de copia de 5 cromosomas, sin profundizar en alteraciones estructurales (es decir, que no afecten al cromosoma completo) o que afecten a otros cromosomas9,10. Por todo ello, son técnicas útiles en poblaciones de gestantes de alto riesgo de presentar síndrome de Down u otras trisomías frecuentes (Edwards, Patau, anomalías de X e Y).
En los últimos años, la incorporación de las tecnologías genómicas y, más en concreto, la de los microarrays de hibridación genómica comparada, abreviados comúnmente como array-CGH, ha supuesto un importante cambio en la rutina del diagnóstico genético. Este impacto es muy evidente en los diagnósticos citogenéticos dirigidos a la detección y análisis de cromosomopatías que desembocan en ganancia o pérdida de material genético11.
Como ventajas frente al cariotipo convencional, el array-CGH permite una mayor resolución en la detección de alteraciones o cambios de número copia (abreviado CNV de copy number variations), pasando de una resolución aproximada de 3-5 megabases en el cariotipo convencional a varias kilobases en el array-CGH. Además, el array permite caracterizar dichas CNV de una manera precisa, conociendo en detalle la localización exacta de la alteración, sus límites, tamaño y el contenido en genes de la misma, lo que redunda en un diagnóstico genético más eficaz y una predicción casi siempre más precisa de la enfermedad asociada a dicha alteración. Como otras ventajas importantes, al contrario que el cariotipo, es importante señalar que el array-CGH no precisa de células en división para la producción de un resultado analizable (pudiendo utilizar fuentes de ADN congelado o incluso fijado); es por tanto una técnica más rápida, similar en respuesta a las técnicas de análisis de aneuploidías12. Las principales desventajas del array-CGH son la imposibilidad de detectar reordenamientos balanceados (algo que sí puede detectar el cariotipo)13 y, debido al aumento de resolución, la aparición de alteraciones no claras, sin explicación clínica aparente, conocidas como alteraciones de significado incierto14.
El array-CGH es, actualmente, una herramienta ampliamente utilizada para la evaluación clínica de pacientes pediátricos, afectos de retraso mental, anomalías congénitas y otros espectros sindrómicos de difícil caracterización15, hasta el punto de que la sociedad americana de medicina genética ha emitido un comunicado recomendando que el array-CGH sea la primera herramienta de utilización en este tipo de pacientes, frente al uso del cariotipo convencional, por las innumerables ventajas que aporta al diagnóstico16.
El uso del array-CGH en el campo de diagnóstico prenatal ha sido comentado y evaluado17, habiendo sido ya publicados algunos estudios que han descrito su uso general18–20, aunque especialmente en aquellas poblaciones de alto riesgo o de malformaciones ecográficas21–23, existiendo menos información acerca del uso del array-CGH para diagnóstico genético prenatal en poblaciones de gestantes de bajo riesgo.
El presente estudio describe el uso de un array-CGH de oligonucleótidos de disen¿ o específico para su uso en diagnóstico prenatal, sobre una población de gestantes de bajo riesgo con la intención de valorar la capacidad de detección de alteraciones genéticas de la técnica (ganancias y/o deleciones), así como comparar esta capacidad con el cariotipo convencional realizado simultáneamente en toda la muestra.
MetodologíaEl array-CGH se ofreció como una opción diagnóstica, adicional al cariotipo convencional, a las gestantes de Policlínica Gipuzkoa sometidas a un procedimiento invasivo de diagnóstico prenatal. Todas las gestantes firmaron un consentimiento informado donde se les informaba acerca de las ventajas y limitaciones de la tecnología utilizada, así como de su uso para fines experimentales.
Este estudio incluye una muestra de 530 gestantes. Para la realización del protocolo de trabajo se extrajeron aproximadamente 15-18ml de líquido amniótico en las semanas 15 a 17. El líquido se repartió en 3 tubos, para realizar, en todos los casos: 2 cultivos celulares para la obtención de metafases y estudio citogenético por cariotipo convencional y una extracción directa de ADN para realizar array-CGH. En caso de no poder obtener suficiente cantidad de ADN directamente a partir del líquido amniótico para realizar el array-CGH, se dio prioridad a los cultivos citogenéticos, pudiendo obtener ADN, posteriormente, a partir de ellos. Asimismo, se informó a los padres de que, en algunos casos, puede resultar necesario realizarles una extracción de sangre para estudio con array, con el fin de confirmar o descartar si un cambio de número de copias encontrado en el array del líquido amniótico era heredado o ex novo, y así poder dar el correspondiente asesoramiento genético.
El estudio del cariotipo convencional con bandas G se realizó siguiendo los protocolos de citogenética prenatal (Cytogenetic Guidelines and Quality Assurance EQA). El array-CGH utilizado por la Unidad de Genética de la Policlínica Gipuzkoa fue KaryoNIM® prenatal, un array-CGH de 15.000 oligonucleótidos, diseñado por la compañía NIMGenetics y fabricado por Agilent Technologies (Santa Clara, CA); dicho array presenta una cobertura general para la detección de cromosomopatías de 2 megabases, así como un enriquecimiento de la resolución en aproximadamente 95 regiones patológicas conocidas (www.nimgenetics.com), detectando en ellas alteraciones de, al menos, 200 kilobases. El protocolo de la técnica array-CGH se realizó siguiendo las especificaciones del proveedor24, utilizando ADN control (Promega) de sexo similar al feto, analizando el sexo del feto bien por ecografía o bien por otras técnicas moleculares complementarias, como FISH.
Los arrays se escanearon con un modelo Agilent a una resolución de 5μm. A partir de las imágenes de fluorescencia escaneadas, se extrajeron los datos de logaritmo de ratio de cada array mediante el software Feature Extraction v10.7 de Agilent Technologies. Esos datos fueron visualizados y analizados mediante el software Agilent Genomic Workbench v. 5.0. A nivel bioinformático, se utilizó un umbral de detección basado en el estadístico ADM-2 de 6, así como un mínimo de 5 sondas consecutivas para considerar una región como patológica. Todos los datos obtenidos fueron codificados en el genoma humano hg19.
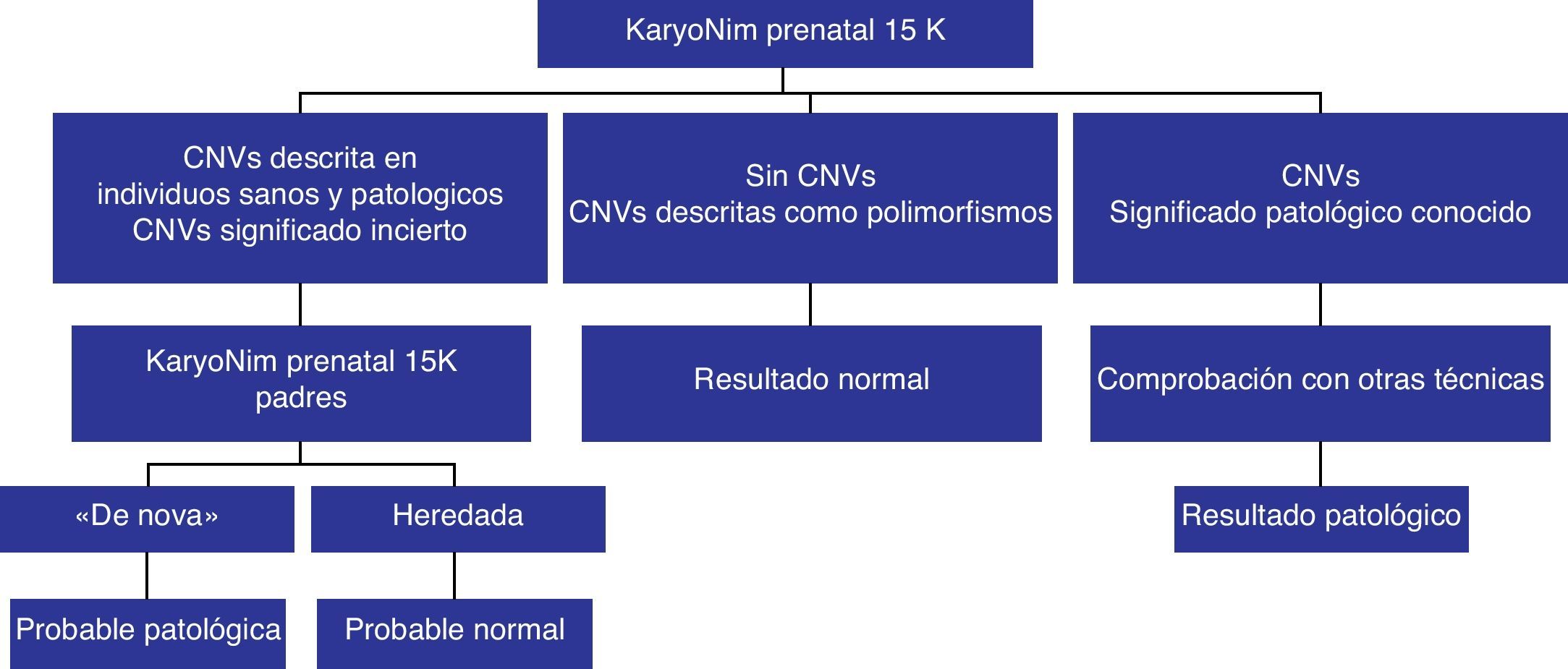
El análisis de los datos obtenidos en cariotipo y array-CGH se realizó de acuerdo a la figura 1, un algoritmo basado en estudios previos15. En los casos en los que no se detectó ninguna CNV o se detectó una CNV descrita como polimorfismo frecuente en la población sana, sin implicación patológica25, se informó con resultado normal. En los casos con detección de una CNV descrita como patológica, se realizó una comprobación posterior, con cariotipo, si era una alteración numérica, o con otras técnicas moleculares, como FISH (Abbott Molecular, Des Plaines, IL, Estados Unidos) o MLPA (MRC Holland, Ámsterdam, Holanda). En los casos con detección de una CNV de significado incierto (desconocimiento de su efecto fenotípico), se realizó un estudio genómico con KaryoNIM® a los progenitores. En el caso de herencia de los progenitores, se informó de que la CNV podría probablemente ser benigna pero que existía el riesgo de penetrancia incompleta o expresividad variable. Si la CNV era ex novo se informó de un riesgo elevado de patogeneicidad para la CNV. En ambos casos se procedió a una confirmación de la CNV por otras técnicas moleculares (FISH o MLPA).

En el caso de que fuera necesario, se realizó un estudio de disomía uniparental mediante amplificación por PCR y posterior electroforesis capilar utilizando un secuenciador Abiprism 3130, visualizando los resultados en el software GeneMapper V4.0 (Life Technologies, Carlsbad, CA, Estados Unidos).
ResultadosEl presente estudio está basado en una muestra total de 530 gestantes sometidas a un procedimiento invasivo de diagnóstico prenatal a las que (como se indica en Material y métodos) se les ofreció el array-CGH como opción diagnóstica adicional al cariotipo convencional. Las indicaciones identificadas para la realización de la amniocentesis fueron: 261 (49%) gestantes presentaban edad materna avanzada (más de 35 años), 253 (48%) gestantes presentaron ansiedad materna sin sospechas patológicas y 16 (3%) presentaban un riesgo superior de embarazos patológicos. De las muestras utilizadas para las hibridaciones con los array-CGH, 242 fueron obtenidas a partir de cultivo celular, y 288 fueron obtenidas directamente a partir de líquido amniótico recién extraído.
La gran mayoría de los casos prenatales analizados, 522 de un total de 530 (98,5%), no presentaron alteraciones de número de copia en el estudio asociadas con enfermedades o síndromes genéticos. En concreto, el 89,5% (474 de 530) de las muestras analizadas presentaron un perfil genómico sin ninguna alteración y el 9% (48 de 530) de las muestras presentaron alteraciones benignas conocidas previamente y sin implicación patológica.
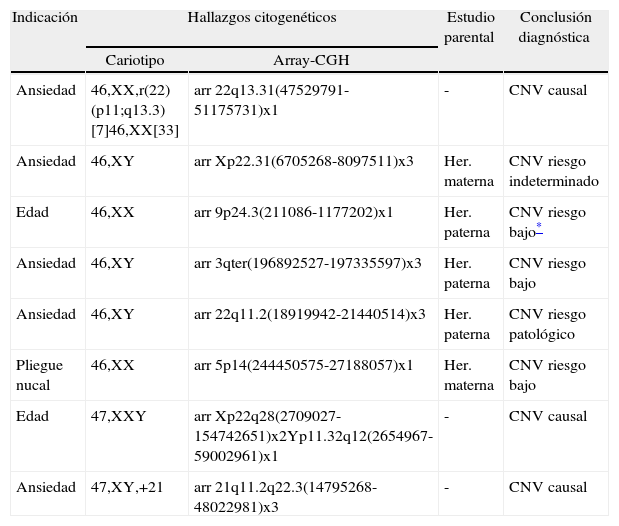
Por otro lado, se detectaron un total de 8 casos (1,5%) con CNV que no aparecen descritas como benignas ni en la literatura ni en las bases de datos genéticas ni genómicas (tabla 1). Mientras que en uno de los casos fue detectado posteriormente un aumento en el tamaño del pliegue nucal, por lo que, en realidad debería ser incluido en la población de alto riesgo, los otros 7 casos mantuvieron su indicación de bajo riesgo (2 por edad materna elevada, 5 por ansiedad materna).
Cambios detectados por array-CGH
| Indicación | Hallazgos citogenéticos | Estudio parental | Conclusión diagnóstica | |
| Cariotipo | Array-CGH | |||
| Ansiedad | 46,XX,r(22)(p11;q13.3)[7]46,XX[33] | arr 22q13.31(47529791-51175731)x1 | - | CNV causal |
| Ansiedad | 46,XY | arr Xp22.31(6705268-8097511)x3 | Her. materna | CNV riesgo indeterminado |
| Edad | 46,XX | arr 9p24.3(211086-1177202)x1 | Her. paterna | CNV riesgo bajo* |
| Ansiedad | 46,XY | arr 3qter(196892527-197335597)x3 | Her. paterna | CNV riesgo bajo |
| Ansiedad | 46,XY | arr 22q11.2(18919942-21440514)x3 | Her. paterna | CNV riesgo patológico |
| Pliegue nucal | 46,XX | arr 5p14(244450575-27188057)x1 | Her. materna | CNV riesgo bajo |
| Edad | 47,XXY | arr Xp22q28(2709027-154742651)x2Yp11.32q12(2654967-59002961)x1 | - | CNV causal |
| Ansiedad | 47,XY,+21 | arr 21q11.2q22.3(14795268-48022981)x3 | - | CNV causal |
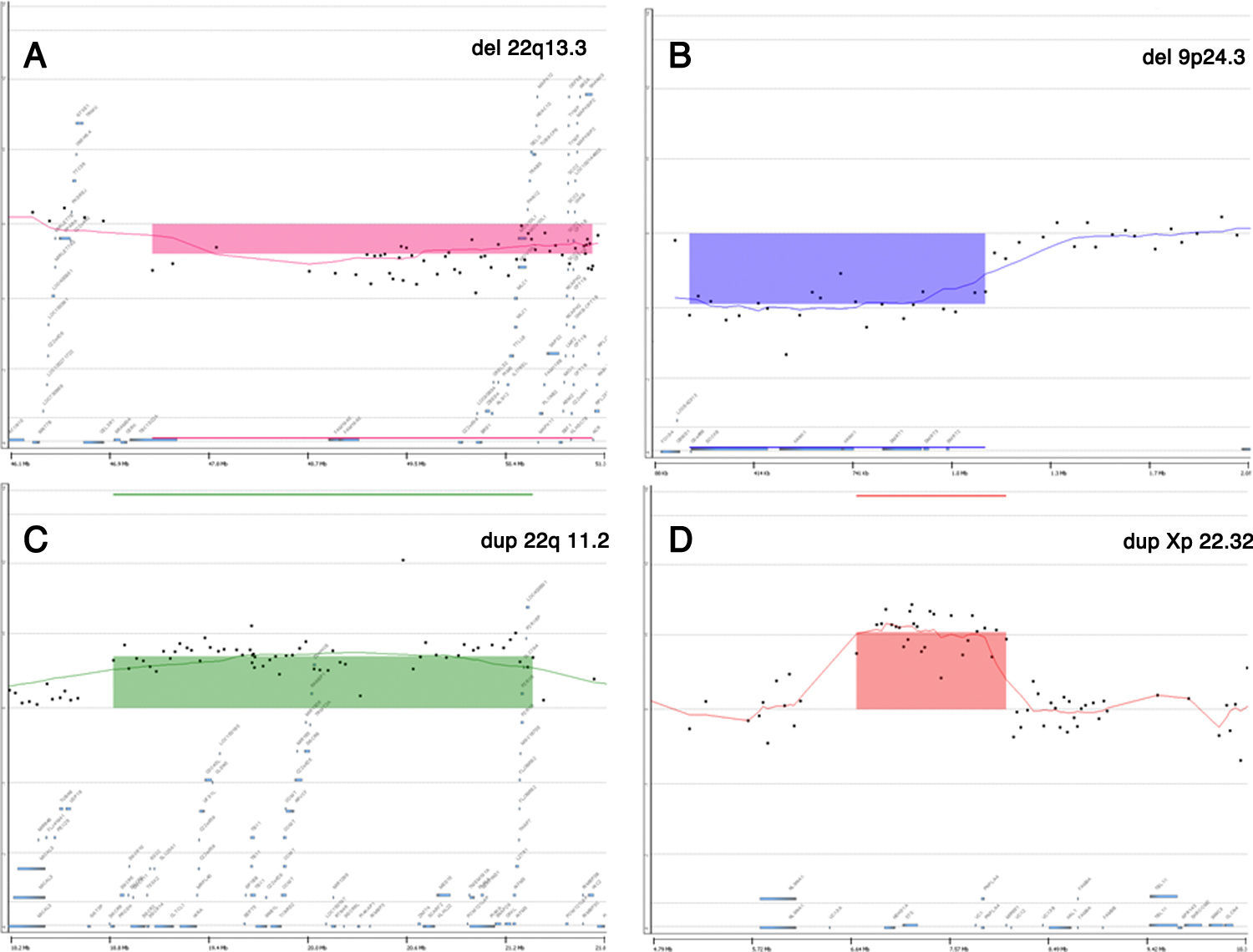
La coincidencia de los resultados de array-CGH y cariotipo convencional (tabla 1) fue obviamente total en los 2 casos con aneuploidías (una trisomía 21 y un síndrome de Klinefelter). Uno de los casos patológicos fue inicialmente identificado mediante el array-CGH como portador de una deleción terminal de aproximadamente 3,78 megabases, afectando la región del síndrome de microdeleción 22q13.3 (OMIM #606232). Posteriormente, el análisis del cariotipo identificó la presencia de un marcador cromosómico en anillo, supuestamente del cromosoma 22, presente en el 15% de las metafases analizadas.
Las CNV alteradas observadas en los 5 casos restantes no pudieron ser identificadas mediante cariotipo convencional. Como se refleja en la tabla 1, las alteraciones presentes en 3 de estos casos fueron identificadas en alguno de los progenitores, por lo que fueron consideradas como variantes heredadas sin asociación patológica conocida y, por lo tanto, informadas como CNV de bajo riesgo. En otro caso, la CNV implicaba una región genómica descrita en la literatura y asociada a un síndrome genético, por lo que se consideró una variante heredada con riesgo patológico. Y finalmente, en un único caso, la CNV identificada también estaba presente en la madre, pero no se pudo asociar a ningún hallazgo o descripción patológica, por lo que retuvo su clasificación como de riesgo indeterminado.
Por otra parte, tras realizar el cariotipo, hubo 3 casos con dotación genómica normal, según el array-CGH, pero portadores de una translocación equilibrada citogenéticamente. Dado que todos los reordenamientos implicaban al cromosoma 14, se procedió a realizar un análisis de disomía uniparental del cromosoma 14, no encontrando ninguna región de homocigosidad para dicho cromosoma.
DiscusiónEn el presente estudio los objetivos fundamentales han sido: 1) comprobar la fiabilidad y la capacidad de detección del array-CGH de alteraciones del número de copia (aneuploidías, ganancias o deleciones) en un conjunto de muestras prenatales con indicaciones de bajo riesgo, comparándolas con el uso del cariotipo convencional; y 2) evaluar la capacidad de detección de cambios de número de copia, tanto detectables por el cariotipo convencional, como aquellos considerados como submicroscópicos, pero sin incrementar el riesgo de detectar alteraciones de significado incierto, debido al diseño específico del array (ver Material y métodos), que pudieran producir dificultades en el diagnóstico prenatal.
Por otra parte, se pretendía estudiar si el uso de array-CGH debería ser restringido, como hasta ahora, a las gestantes de alto riesgo (como segunda opinión tras cariotipo) o podría ser utilizado como técnica de primera línea frente al uso del cariotipo.
El estudio genético utilizando array-CGH detectó todas las CNV que podrían haber sido detectables por cariotipo convencional (tanto la trisomía del cromosoma 21, como el caso XXY), así como identificó un cambio citogenético visible en un mosaico de bajo porcentaje (15%), pero cuya complejidad hacía difícil su correcta identificación por cariotipo, e imposible, definir exactamente el tamaño delecionado (fig. 2A). Así mismo, detectó 5 alteraciones no visibles por cariotipo convencional y no incluidas en los listados de polimorfismos genéticos sin asociación patológica. Es importante señalar que todos los resultados de array-CGH fueron validados posteriormente bien por cariotipo, FISH, MLPA, o fueron comprobados al presentar, alguno de sus progenitores, la misma CNV, lo que demuestra que no existió ningún falso positivo descrito en nuestro estudio.
 Deleción en la citobanda 22q13. B) Deleción en la citobanda 9p24.3. C) Duplicación en la citobanda 22q11.2. D) Duplicación en la citobanda Xp22.32.")
El array-CGH no detectó alteraciones citogenéticas sin CNV asociadas en tres casos: dos translocaciones t(13;14), una ex novo y otra heredada, y una translocación t(14;22) ex novo en un tercer caso (0,5% casos del total). A pesar de que el array-CGH es incapaz de detectar translocaciones equilibradas, sí puede identificar si concurren alteraciones de cambio de número de copia asociadas a las mismas, invisibles al cariotipo26. De hecho, se ha demostrado que las translocaciones equilibradas ex novo que rompen un gen con consecuencia fenotípica son un evento extremadamente raro en la población humana (cercano a 1 entre 10.000)27. Adicionalmente, se decidió completar el estudio en estos casos realizando un análisis de UPD en el cromosoma 14, descartando la homocigosidad para dicho cromosoma en los tres casos.
El presente estudio detectó 5 fetos con CNV que no eran claramente patológicas, pero tampoco se encontraban en zonas claramente benignas. Según el algoritmo utilizado, tres de ellas se consideraron CNV heredadas de bajo riesgo. No obstante, una de esas tres CNV fue exhaustivamente estudiada por razones de consejo genético. Se trataba de un feto de sexo femenino que presentaba una deleción en 9p24.3, de aproximadamente una megabase, heredada del padre (fig. 2B). El estudio molecular posterior identificó dicha deleción, además de en el feto, en el padre y el hermano nacido previamente. La deleción de ese segmento, incluyendo los genes DOCK8, DMRT1, DMRT3 y DRMT2, se asocia bibliográficamente con el síndrome OMIM (#154230) conocido como Disgenesia gonadal 46,XY completa o parcial, asociada a deleción 9p24.328. A pesar de que el actual embarazo se había conseguido de manera espontánea, al estudiar los antecedentes familiares para realizar el asesoramiento genético, se comprobó que el hijo previo de la pareja había sido concebido por métodos de reproducción asistida debido a la infertilidad de la pareja, lo que podría indicar una infertilidad debida a una posible disgenesia gonadal parcial presente en el padre. Tras el asesoramiento genético la pareja decidió continuar el embarazo, teniendo en cuenta que la ecografía de alta resolución que se les aconsejó realizar era normal y además que el feto era 46,XX.
De las otras 2 alteraciones restantes, una fue catalogada como CNV heredada de alto riesgo, ya que era una duplicación 22q11.2 de 2,5 megabases (fig. 2C), posteriormente identificada en un progenitor sano (padre). Esta alteración está asociada clínicamente al síndrome de duplicación 22q11.2 (OMIM #608363), una entidad clínica compleja29,30 siendo descrita como de fenotipo variable y penetrancia incompleta, lo que resulta en la exitencia de individuos portadores de la duplicación tanto sanos como afectos. Patológicamente los pacientes con dicha duplicación podrían presentar retraso del desarrollo, hipotonía, dismorfias leves y otros síntomas menores. Sin embargo, la bibliografía hace necesario expresar que muchas personas portadoras de la duplicación eran sanas sin incidencia patológica. Tras realizar un consejo genético al respecto a los progenitores, estos decidieron continuar el embarazo al presentar el feto la misma duplicación que el progenitor sano.
La última CNV descrita fue una CNV de significado incierto, duplicación Xp22.32 (fig. 2D), heredada de la madre sana, de 1,3 megabases, encontrada en un feto de sexo masculino. Dicha duplicación afectaba la región de la ictiosis ligada al X (OMIM #308100), incluyendo al gen STS, existiendo numerosas citas bibliográficas que la describen31–33. Según dichas fuentes, esta duplicación se encuentra presente en el 0,15% de la población sana y en el 0,37% de individuos con fenotipo patológico que presentan discapacidades intelectuales y de comportamiento, siendo generalmente heredada de un progenitor. Las publicaciones señaladas no pueden concluir una asociación real entre la duplicación y la enfermedad, hasta el punto de que una de ellas la considera una variante benigna31, mientras que otra la considera un factor de riesgo33. Después de informar a la pareja en el correspondiente consejo genético, decidieron continuar el embarazo.
Actualmente, las bases de datos públicas de CNV polimórficas y patológicas, así como los estudios recientes realizados en array-CGH son lo suficientemente completos para poder dar un consejo genético adecuado en el diagnóstico prenatal, en el caso de encontrar CNV sospechosas de tener una asociación patológica25,34,35. Asimismo, es extremadamente importante realizar un consejo genético adecuado, que permita explicar a los progenitores el significado de los resultados genéticos obtenidos36.
El uso del array-CGH como herramienta de primera línea, a nivel prenatal, está siendo actualmente debatido en la comunidad científica37. Los resultados del presente estudio, al combinarse con experiencias previas, indican que el array-CGH es una herramienta eficaz, que puede ser utilizada en la rutina del diagnóstico prenatal, informando de su capacidad de detección y limitaciones, aunque por ahora se recomienda realizar un estudio paralelo de cariotipo, al ser esta la técnica más extendida a nivel clínico.
En conjunto, los resultados extraídos del presente estudio hacen ver que el array-CGH es capaz de detectar los eventos patológicos que podrían haber sido detectables mediante cariotipo, clarificando las alteraciones complejas y reduciendo el riesgo de informar reordenamientos posiblemente no equilibrados.
Por todo ello, aunque es necesario seguir realizando cohortes de mayor tamaño para poder establecer la capacidad del array-CGH en el diagnóstico prenatal, nuestros datos, junto con los de otros estudios previos, permiten realizar una valoración positiva del array-CGH como herramienta de diagnóstico en el uso prenatal de poblaciones de bajo riesgo.
Conflicto de interesesJavier Suela y Juan Cruz Cigudosa son, respectivamente, director técnico y consultor de NIMGenetics, laboratorio privado especialista en el análisis genómico para el diagnóstico clínico y la investigación.
A los doctores J.C. Trecet, E. Otarola, M. Gabarain y J. Rodríguez, de la Policlínica Gipuzkoa, por su colaboración en el estudio.