



O higroma quístico (HQ) é uma malformação congénita diagnosticada durante a gravidez pela demonstração ecográfica de uma estrutura quística na região occipitocervical. Pode surgir isolado ou em associação a anomalias cromossómicas, malformações fetais ou síndromes genéticas, com prognóstico global reservado.
O objetivo deste trabalho foi avaliar a conduta obstétrica e os resultados pediátricos dos sobreviventes com higroma quístico (HQ).
Material e métodosEstudo retrospetivo descritivo de 224 grávidas com HQ fetal, diagnosticadas ou referenciadas ao Centro de Diagnóstico Pré-Natal da nossa instituição, entre janeiro de 1991 e julho de 2011.
ResultadosA idade gestacional média ao diagnóstico foi de 13 semanas, 77,7% dos casos diagnosticados no primeiro trimestre. Ecograficamente, 66 casos estavam associados a hidrópsia. O cariótipo fetal foi determinado em 206 casos, com deteção de 107 anomalias cromossómicas. O cariótipo foi normal em 99 casos, tendo sido detetados 12 casos de doenças genéticas e 18 de malformações estruturais fetais. A interrupção médica de gravidez foi a opção de 111 pacientes, registaram-se 39 casos de morte in utero e 61 nados-vivos. O tempo médio de seguimento dos sobreviventes foi de 75 meses, tendo-se verificado um desenvolvimento psicomotor adequado em 30 casos.
ConclusãoPerante o diagnóstico de HQ, é essencial esclarecer a etiologia, de modo a definir o prognóstico e orientar corretamente a gravidez. Existe uma forte correlação entre o diagnóstico de HQ com aneuploidia fetal, conferindo-lhe pior prognóstico em comparação com os casos de HQ sem evidência de alterações cromossómicas ou malformações estruturais fetais, geralmente com bons resultados neonatais e pediátricos.
Fetal cystic hygroma (CH) is a congenital malformation prenatally diagnosed by the demonstration of a cystic structure in the occipitocervical region on ultrasound. It may appear isolated or in association with chromosomal abnormalities, fetal malformations or genetic syndromes, with a poor overall prognosis.
The main purpose of this work was the evaluation of the obstetric management and paediatric outcome for the survivors of CH.
Material and methodRetrospective analysis of 224 pregnant women with fetal CH, diagnosed or referred to our prenatal diagnosis centre, from January 1991 to July 2011.
ResultsThe mean gestational age at diagnosis was 13 weeks and 77.7% of cases were diagnosed in the first trimester. On ultrasound, 66 cases were associated with hydrops. Fetal karyotype was obtained in 206 cases, and chromosomal abnormalities were found in 107. Fetal karyotype was normal in 99 cases, detected 12 cases of genetic diseases and 18 cases of fetal malformations. Elective pregnancy termination was undertaken by 111 patients. There were 39 cases of spontaneous fetal demise and 61 live births. The mean follow-up of survivors was 75 months, and normal paediatric outcome was confirmed in 30 cases.
ConclusionIt is essential to clarify the underlying aetiology of CH in order to establish a prognosis and counselling. There is a strong association with fetal aneuploidy and significantly worse outcome in contrast in cases without evidence of chromosomal or structural abnormalities, most of them carrying good prognosis.
A avaliação fetal ecográfica no primeiro trimestre (1.° T) envolve a medição da translucência da nuca (TN), um pequeno espaço hipoecogénico na região do pescoço, resultante de edema subcutâneo. Um aumento excessivo deste espaço pode estar relacionado com o aumento da TN ou com higroma quístico (HQ), 2 entidades associadas ao aumento do risco de anomalias cromossómicas, malformações fetais ou síndrome genéticas1,2. Para alguns autores, como Nicolaides, estas 2 entidades fazem parte de um espectro patológico contínuo com diferentes graus de envolvimento fetal3. No entanto, este conceito não é consensual e, para a maioria dos autores, trata-se de 2 diagnósticos diferentes, com um significado clínico distinto, tendo o HQ pior prognóstico do que o simples aumento da TN4.
O HQ é uma malformação congénita do sistema linfático em que ocorre uma obstrução na sua comunicação com o sistema venoso, com consequente acumulação de linfa nos sacos jugulares linfáticos na região da nuca1.
A sua prevalência é de 1 em 100 fetos no 1.° T da gravidez1 e de 1 em 6000 nascimentos quando a gestação é levada a termo5.
O diagnóstico é ecográfico e ocorre principalmente no 1.° T, consistindo numa estrutura quística anecoica, simples ou septada, localizada mais frequentemente na região occipitocervical1.
O diagnóstico diferencial é realizado com defeitos fetais do tubo neural, como encefalocelo posterior, meningocelo cervical, teratoma quístico e hemangioma, sendo que, no HQ, o crânio e coluna estão intactos, não tem conteúdo sólido, pode apresentar septos internos e a sua vascularização é reduzida1,6.
O risco de anomalias cromossómicas associado ao HQ é cerca de 50%, sendo mais frequentes a síndrome de Turner (monossomia X) e as trissomias (21,18,13)1, com predomínio das trissomias nos HQ diagnosticados no 1.° T3,4,7 e da monossomia X naqueles diagnosticados no segundo trimestre (2.° T)1,8. O risco de aneuploidia parece, segundo alguns estudos, estar aumentado nos HQ septados9,10 e invariavelmente com o aumento da TN1,3. Segundo Kogan et al., o risco de anomalias cromossómicas é de cerca de 20% para uma TN entre 3,5-4,4mm, de 50% para uma TN entre 5,5-6,4mm e de 75% para uma TN>8,5mm, verificando-se ainda que a maioria dos fetos com trissomia 21 apresenta uma TN<4,5mm e que aqueles com trissomia 13, 18 e síndrome de Turner têm maioritariamente valores de TN>4,5mm11. Segundo outros autores, neste último grupo de aneuploidias destaca-se a monossomia X por estar associada a valores de TN superiores3,4.
Cerca de um terço dos fetos euploides com HQ apresentam malformações estruturais, sobretudo cardíacas, mas também hérnias diafragmáticas, anomalias renais, defeitos da parede abdominal, anomalias ósseas e síndromes polimalformativos1,4,7,12. A incidência de malformações fetais parece estar aumentada nos HQ septados4 e a prevalência das malformações cardíacas está relacionada com o aumento da espessura da TN12,13.
Entre 0,5 a 6,5% dos HQ em fetos euploides associam-se a síndromes genéticas1. Foram descritos cerca de 100 síndromes, algumas relativamente raras, entre elas a síndrome de Noonan, a síndrome Pterygium múltiplos, a díndrome de Roberts, a síndrome de Cornelia de Lange, hiperplasia congénita adrenal, atrofia muscular espinhal, síndrome DiGeorge, síndrome Smith-Lemli-Opitz e várias displasias ósseas1,5,12.
Relativamente ao prognóstico, é de esperar um prognóstico pediátrico normal em 15 a 30% dos fetos com HQ, sendo o risco de morte fetal ou neonatal nos fetos euploides de cerca de 25%1. Os fatores de mau prognóstico incluem a aneuploidia, presença de malformações estruturais fetais e marcadores ecográficos, como a presença de septos internos, persistência do higroma, associação de hidrópsia e espessura da TN1,12. O prognóstico específico de cada feto depende dos achados associados ao HQ, sendo, de um modo geral, os resultados favoráveis quando o cariótipo é normal e na ausência de malformações estruturais associadas, verificando-se nestes casos 80% de regressão espontânea do higroma e crianças, na sua maioria fenotipicamente normais8. Houve, no entanto, alguns estudos que reportaram um aumento da prevalência de um atraso de desenvolvimento cognitivo em cerca de 2-3,9% destes casos12. Perante situações de aneuploidia, malformações estruturais graves, associação ou evolução para hidrópsia, o prognóstico é reservado e associado a elevada mortalidade1.
O significado do HQ na gravidez múltipla depende da corionicidade. Nas gestações dicoriónicas, as implicações do HQ são semelhantes às da gravidez unifetal, sendo possível o feticídio seletivo, nos casos em que está indicado e é desejado. Nas gestações monocoriónicas, a avaliação da TN não tem a mesma fiabilidade pela grande taxa de falsos positivos, embora continue a ser um marcador preditivo importante de anomalias cromossómicas e da ocorrência malformações estruturais fetais1,2.
Relativamente à conduta obstétrica, perante o diagnóstico de HQ é fundamental esclarecer a etiologia e detetar as possíveis anomalias associadas. Deste modo, deve ser determinado o cariótipo fetal, pelo risco subjacente de anomalias cromossómicas, e realizada ecografia morfológica detalhada precoce e ecocardiografia fetal para deteção de malformações estruturais fetais. Pelo risco aumentado de morte fetal intrauterina e perinatal, é fundamental realizar uma avaliação ecográfica seriada do crescimento fetal e monitorização precoce do bem-estar materno-fetal no terceiro trimestre (3.° T)1. Salienta-se ainda a importância de uma vigilância mais cuidada da saúde materna, nomeadamente em situações raras que podem surgir num quadro de HQ associado a hidrópsia, como o Mirror Syndrome, um quadro clínico que se pode apresentar de forma semelhante a uma pré-eclâmpsia grave14. O aconselhamento genético é essencial ao longo de todo o processo para a correta orientação da gravidez atual e avaliação das implicações em gravidezes futuras, pelo risco de recorrência de algumas doenças associadas ao HQ4,6.
O objetivo deste estudo foi a avaliação das características diagnósticas e conduta obstétrica das gestações com o diagnóstico fetal de HQ, bem como a determinação da morbilidade neonatal e pediátrica associada.
Material e métodosFoi efetuado um estudo retrospetivo descritivo de 224 grávidas com o diagnóstico fetal de HQ, diagnosticadas ou referenciadas ao Centro de Diagnóstico Pré-Natal da nossa instituição, no período compreendido entre janeiro de 1991 e julho de 2011.
O diagnóstico fetal de HQ foi realizado mediante a determinação ecográfica, transvaginal ou transabdominal, no plano transverso e médio-sagital na região da nuca, de uma estrutura quística hipoecogénica ou anecoica, simples ou septada, de localização occipitocervical7,15.
Perante o diagnóstico de HQ, foi proposto por rotina a determinação do cariótipo fetal (amniocentese ou biópsia das vilosidades coriónicas) e oferecido aconselhamento genético. Foi realizada uma ecografia cerca de 4 semanas após o diagnóstico de HQ, para documentar a progressão ou regressão do higroma, assim como a associação de hidrópsia fetal. O diagnóstico de hidrópsia foi considerado quando se verificou a presença de edema subcutâneo associado a derrame pleural, pericárdico e/ou ascite13.
Foi efetuada uma ecografia morfológica detalhada precoce para deteção de malformações estruturais fetais e realizada ecocardiografia fetal entre as 18 e 20 semanas. Nas gravidezes em evolução foi realizada avaliação ecográfica seriada do crescimento fetal e monitorização precoce do bem-estar fetal.
Procedeu-se ao registo dos resultados obstétricos e neonatais (morte in utero, interrupção médica de gravidez, interrupção voluntária de gravidez, nados-vivos e morbilidade neonatal) e obteve-se um follow-up pediátrico dos sobreviventes durante um período de 75 (9-193) meses, através da avaliação dos registos médicos dos geneticistas pediátricos ou pediatras ou informação dada pelos pais mediante contacto telefónico.
O programa estatístico aplicado foi o Statistical Package for Scientific Science (SPSS) versão 15.0. Foi utilizado o teste chi-square (χ2) para comparar variáveis categóricas e o teste t-student para variáveis numéricas, tendo-se considerado p<0,05 com significado estatístico.
ResultadosDurante o período do estudo foram diagnosticados 224 casos de higroma quístico fetal, 4 destes em gravidezes múltiplas (3 monocoriónicas e uma bicoriónica). A idade materna média na data do diagnóstico foi de 30,3±5,8 (14-44) anos e 53,5% das mulheres eram nulíparas. Dos antecedentes salienta-se um caso com história prévia de 2 gestações anteriores com o diagnóstico de HQ. A idade gestacional média ao diagnóstico foi de 12,9±3,0 (9-34) semanas, sendo que 77,7% dos casos foram diagnosticados no 1.° T (< 14 semanas), manifestando-se os restantes apenas no 2.° T.
Ecograficamente, a espessura média do HQ foi de 5,1±2,2 (2-12) mm, tendo sido classificado como septado em 17 (7,6%) casos e detetada hidrópsia fetal associada no momento do diagnóstico inicial em 66 (29,5%).
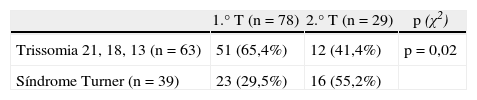
O cariótipo fetal foi determinado em 206 casos: 31 por biópsia das vilosidades coriónicas (BVC), 174 por amniocentese e um por cordocentese. Foram detetadas anomalias cromossómicas em 107 (51,9%) casos, sendo a aneuploidia mais comum a síndrome de Turner (n=39; 36,4%), seguida da trissomia 21 (n=31; 29%), da trissomia 18 (n=24; 22,4%), da trissomia 13 (n=8; 7,5%), das translocações cromossómicas equilibradas (n=4; 3,7%), com o registo de um caso de 69, XXX (n=1; 0,9%). Verificou-se um predomínio das trissomias nos HQ diagnosticados no 1.° T (65,4%) e da síndrome de Turner no 2.° T (55,2%), com significado estatístico (tabela 1).
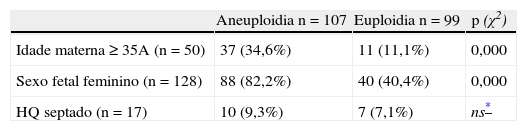
As variáveis idade materna igual ou superior a 35 anos, sexo fetal feminino e valores de TN mais elevados associaram-se de forma estatisticamente significativa à presença de anomalias cromossómicas. Apesar de os HQ septados serem mais frequentes nos fetos com aneuploidia, este dado não foi estatisticamente significativo (tabela 2).
Cariótipo fetal (n=206)
| Aneuploidia n=107 | Euploidia n=99 | p (χ2) | |
| Idade materna ≥ 35A (n=50) | 37 (34,6%) | 11 (11,1%) | 0,000 |
| Sexo fetal feminino (n=128) | 88 (82,2%) | 40 (40,4%) | 0,000 |
| HQ septado (n=17) | 10 (9,3%) | 7 (7,1%) | ns* |
| Aneuploidia n=107 | Euploidia n=99 | p (t-student) | |
| Espessura TN (mm) | 6,5±2,6 (2-12) | 4,3±1,6 (1,3-9) | 0,001 |
Analisando a relação entre a espessura da TN com o tipo de aneuploidia, não se verificaram diferenças significativas, contrariamente ao descrito na literatura, sendo a TN ≥ 4,5 na maioria das anomalias cromossómicas (86,4%). Verificámos, no entanto, valores de TN médios superiores para a monossomia X em relação às restantes aneuploidias (8,1±4,2; 2-12 vs. 6,1±1,9; 2,3-9,3).
Foram detetadas ecograficamente 18 malformações estruturais fetais (13 cardíacas, 3da parede abdominal, uma anomalia renal e outra óssea), todas em HQ simples, na sua maioria em fetos com aneuploidia (8/13) e sem relação estatística com a espessura da TN (p>0,05).
Relativamente ao diagnóstico etiológico, verificou-se que a causa mais frequente subjacente ao higroma quístico foram as alterações cromossómicas (107;47,8%), seguidas de 12 (5,3%) doenças genéticas (9 em fetos euploides) (tabela 3), 3 (1,3%) cardiopatias graves, 4 (1,8%) síndromes polimalformativos, um (0,5%) caso de infeção por Parvovírus B19 e 79 (35,3%) casos considerados idiopáticos, após investigação protocolada sem alterações. Em 18 (8%) casos não se encontrou uma causa subjacente, pela ausência de estudo, maioritariamente em situações de abortos do 1.° T ou por perda de follow-up.
Nas gravidezes evolutivas verificou-se ecograficamente regressão espontânea do higroma em 72 casos (91,3% fetos euploides, 93% HQ simples, 98,6% sem hidrópsia inicial), evolução para hidrópsia em 19 e agravamento de 33 situações já com o diagnóstico inicial de hidrópsia.
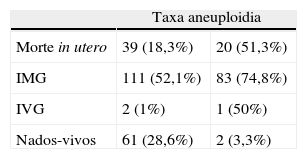
Os resultados obstétricos disponíveis para 213 gestações, excluídas 11 mulheres perdidas para follow-up, estão descritos na tabela 4, podendo verificar-se que houve um mau desfecho obstétrico (morte in utero, interrupção médica de gravidez – IMG, interrupção voluntária de gravidez – IVG) em 152 (71,4%) casos, com 61 (28,6%) nados-vivos.
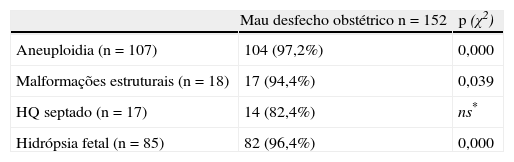
Após a análise estatística dos resultados, constatou-se que a presença de aneuploidia, de malformações estruturais fetais e marcadores ecográficos como a persistência do higroma associado a hidrópsia e espessura da TN, se associam a pior prognóstico (p<0,05) (tabela 5). Não se verificou associação estatisticamente significativa aos HQ septados.
Fatores de mau prognóstico
| Mau desfecho obstétrico n=152 | p (χ2) | |
| Aneuploidia (n=107) | 104 (97,2%) | 0,000 |
| Malformações estruturais (n=18) | 17 (94,4%) | 0,039 |
| HQ septado (n=17) | 14 (82,4%) | ns* |
| Hidrópsia fetal (n=85) | 82 (96,4%) | 0,000 |
| Mau desfecho obstétrico n=152 | p (t-student) | |
| Espessura TN (mm) | 6,1±2,3 (2-12) | 0,001 |
Relativamente aos resultados pós-natais dos 61 nados-vivos, em 13 casos perdeu-se o follow-up (partos ocorridos fora da nossa instituição), tendo os restantes 48 mantido vigilância periódica em consulta de genética pediátrica ou noutra subespecialidade, caso a patologia o justificasse, durante um tempo médio de 75,5±55,4 (9-193) meses. Registaram-se 30 (30/48, 62,5%) casos com um desenvolvimento psicomotor adequado e 18 (18/48, 37,5%) casos de crianças com patologia (tabela 6).
DiscussãoDesde o final dos anos 80 que a determinação da TN assumiu um papel fundamental no rastreio das cromossomopatias, o que foi essencial para o aumento da deteção e em fases mais precoces do HQ fetal, como pudemos constatar na nossa amostra, já que a maioria (77,7%) dos HQ foi diagnosticada no 1.° T2,3.
Existe, segundo a literatura, uma forte correlação entre o HQ e a aneuploidia, confirmada no nosso estudo, no qual foram detetadas 51,9% de anomalias cromossómicas, maioritariamente a síndrome de Turner (36,4%). Verificámos um predomínio estatisticamente significativo do diagnóstico deste tipo de aneuploidia no 2.° T de gestação e das trissomias (21,18,13), no 1.° T. Este facto pode ser explicado, segundo Johnson et al., por este tipo de aneuploidia, ligada a um cromossoma sexual, ter por base mecanismos desencadeadores mais tardios e por as aneuploidias não ligadas aos cromossomas sexuais se associarem a abortos mais precoces antes das 16-20 semanas8.
A idade média materna foi significativamente superior (≥ 35anos) no grupo de fetos com aneuploidias, o que se pode explicar pela maior frequência de anomalias cromossómicas nesta faixa etária. Verificámos ainda uma discrepância do sexo fetal entre os grupos de fetos euploides e aqueles com aneuploidia, com predomínio do sexo fetal feminino no último, o que poderá ser justificado pelo diagnóstico maioritário de síndrome de Turner, neste grupo de fetos7.
Também os nossos resultados foram consistentes com os da literatura ao constatar a associação entre o risco de aneuploidia e o aumento da espessura da TN, o que não se verificou com os HQ septados. Este facto pode provavelmente refletir a falta de uma definição standard em relação ao seu diagnóstico ecográfico, o que justificaria o baixo número de casos de HQ septados da nossa amostra. Relativamente ao tipo de aneuploidia, não encontrámos diferenças estatisticamente significativas com a espessura de TN, embora se tenha verificado, como já descrito anteriormente, uma tendência para valores de TN médios superiores na monossomia X, em relação às restantes aneuploidias.
As malformações estruturais detetadas ecograficamente foram predominantemente cardíacas (13/18) e apenas 8 foram diagnosticadas em fetos euploides (8,1%), valor inferior ao descrito na literatura (um terço dos fetos euploides)1. Também a presença de malformação estrutural fetal não se associou à presença de septos internos nem à espessura da TN4,12,13. A incidência de doenças genéticas no grupo de fetos euploides da nossa amostra (12,1%) foi superior à descrita noutros estudos (0,5 a 6,5%), tendo sido diagnosticados 12 síndromes genéticos com associação conhecida com HQ.
Etiologicamente, destaca-se um caso de infeção materno-fetal por Parvovírus B19, a única infeção já descrita em associação ao aumento da espessura da TN, atribuída a disfunção miocárdica ou anemia fetal por supressão da hematopoiese12.
Quanto à evolução ecográfica das gravidezes evolutivas, registou-se a regressão espontânea do HQ em 72 casos, maioritariamente nas situações de melhor prognóstico, nomeadamente em fetos euploides, HQ sem septos internos, tendo esta ocorrido em apenas um dos casos com o diagnóstico inicial de hidrópsia. Nestes casos de regressão do HQ, ocorre provavelmente recanalização entre o sistema venoso e linfático ou criam-se vias colaterais alternativas de drenagem da linfa9,10. As situações de evolução para hidrópsia ou agravamento de hidrópsia inicial ocorreram principalmente nos casos de aneuploidia e HQ septados, fatores associados a um pior desfecho.
Analisando os resultados obstétricos disponíveis para as 213 gestações com follow-up, verificou-se um mau desfecho obstétrico em 152 (71,4%) casos, tendo-se registado morte in utero em 39 (18,3%), 9 em fetos euploides (9,1%), valor inferior ao da literatura (25%), o que poderá estar influenciado pela decisão parental de realizar IMG em 111 (52,1%) situações e IVG em 2 (1%), mesmo em fetos sem fatores de mau prognóstico associados conhecidos, entre eles, a presença de anomalias cromossómicas1. A análise estatística da nossa amostra permitiu definir, com significado estatístico, como fatores de mau prognóstico para um mau desfecho obstétrico (morte in utero, IMG, IVG), a aneuploidia, a presença de malformação estrutural fetal, a associação de hidrópsia e valores superiores de TN. A presença de septos internos, na sua fisiopatologia relacionada com uma obstrução linfática completa e uma incidência superior de aneuploidia e malformações estruturais fetais, está, segundo a literatura, associada a pior desfecho obstétrico10. No entanto, na nossa amostra, não revelou associação prognóstica estatisticamente significativa, embora se tenha verificado uma predominância deste fator nestes casos.
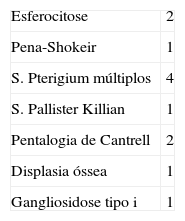
Registaram-se 61 nados-vivos (28,6%), tendo-se perdido o follow-up em 13 casos. Muitos estudos anteriores apresentam a limitação da ausência de resultados pós-natais a longo prazo. No nosso estudo, manteve-se a vigilância de 48 casos durante um tempo médio de 75,5±55,4 (9-193) meses. Constatou-se um desenvolvimento psicomotor adequado em 30 casos (30/224, 13,4%), valor inferior ao encontrado na literatura (15-30%), mas com a limitação de se ter perdido informação em 11 gestações e em 13 nados-vivos1. Foram ainda documentados 18 casos de patologia, predominantemente cardíaca, destacando-se 4 casos de síndrome de Noonan, frequentemente associado a HQ, e um caso raro de síndrome de Aarskog, uma doença genética ligado ao cromossoma X. No nosso trabalho, todos os casos com patologia foram diagnosticados precocemente; no entanto, alerta-se para a possibilidade da existência de doenças com manifestações tardias e da importância de manter uma vigilância pós-natal a longo prazo6. Registou-se ainda um óbito numa criança de um ano de idade com esferocitose.
Assim, perante a deteção de HQ, é fundamental estabelecer-se uma estratégia diagnóstica estruturada, acompanhada por uma conduta direcionada e um aconselhamento genético faseado. Destaca-se a necessidade de esclarecer sobre o risco de aneuploidia após o diagnóstico ecográfico de HQ alertar para a possibilidade de malformações estruturais fetais, doenças genéticas e morte in utero em fetos com cariótipo normal e, por fim, após a ausência de evidência do diagnóstico destas situações, poderá esperar-se um desenvolvimento pediátrico normal em 95% dos casos4. O aconselhamento genético em gestações futuras pode ser pouco importante em situações como a síndrome de Turner ou aneuploidias com baixo risco de recorrência, mas é fundamental nas situações de diagnóstico de doenças monogénicas, muitas vezes com um elevado risco de recorrência associado. Deste modo, a vigilância ecográfica precoce e a determinação do cariótipo fetal são uma conduta aceitável na tranquilização do casal6.
Por fim, podemos concluir que, perante o diagnóstico de HQ, é essencial esclarecer a etiologia subjacente, de modo a definir o prognóstico e proceder à correta orientação da gravidez. A forte correlação entre HQ e aneuploidia confere-lhe pior prognóstico, assim como situações de malformações estruturais fetais graves e de hidrópsia associadas a elevada mortalidade, em comparação com os casos de HQ sem evidência de alterações cromossómicas ou malformações estruturais fetais, em que os resultados são geralmente favoráveis e as crianças, na sua maioria, fenotipicamente normais.
Responsabiliades éticasProteção de pessoas e animais. Os autores declaram que para esta investigação não se realizaram experiências em seres humanos e/ou animais.
Confidencialidade dos dados. Os autores declaram que não aparecem dados de pacientes neste artigo.
Direito à privacidade e consentimento escrito. Os autores declaram que não aparecem dados de pacientes neste artigo.
Conflito de interessesOs autores declaram não haver conflito de interesses.
Os resultados parciais deste trabalho foram apresentados sob a forma de poster no XXII European Congress on Perinatal Medicine, em Granada, de 26 a 29 de Maio de 2010, com o título: Cystic hygroma – Experience of 18 years of our department. department.