


Presentamos el caso de una gestante en la cual se lleva a cabo el diagnóstico de hidrops fetal, a las 17 semanas de embarazo, en dos gestaciones consecutivas. Descartada la etiología inmune, en la segunda gestación se investigó –entre otras– la asociación con enfermedades de depósito lisosomal, detectándose una actividad disminuida de la beta-glucuronidasa en amniocitos cultivados y perfil de glucosaminoglucanos en líquido amniótico, indicativos de enfermedad de Sly o mucopolisacaridosis tipo VII, de herencia autosómica recesiva.
We report the case of a pregnant woman who was diagnosed with hydrops fetalis at 17 weeks in two consecutive pregnancies. Once an immune origin was ruled out, the association with lysosomal storage diseases was investigated during the second pregnancy. This showed decreased activity of beta-glucuronidase in cultured cells and a glycosaminoglycan profile in amniotic fluid, indicative of Sly disease or mucopolysaccharidosis type VII which is inherited in an autosomal recessive pattern.
El hidrops fetal o acúmulo anormal de líquido seroso en al menos dos compartimentos fetales (edema subcutáneo, derrame pericárdico, hidrotórax o ascitis) puede ser el resultado de una gran variedad de etiologías, incluyendo –entre otras– cromosomopatías, síndromes monogénicos, cardiopatías estructurales, arritmias fetales, afección pulmonar, anomalías gastrointestinales, anomalías del tracto genitourinario, anomalías con alto gasto cardiaco (aneurisma de la vena de Galeno, teratoma sacrococcígeo, corioangioma placentario, etc.), anomalías hematológicas o infecciones.
En la actualidad el mecanismo inmunológico (debido a la presencia de anticuerpos maternos contra antígenos eritrocitarios fetales) representa el 10% de los casos y el hidrops no inmune es responsable del 75-87%; por etiologías, las enfermedades metabólicas hereditarias constituyen el 0,5-1,1%, entre las cuales destaca la presencia de las enfermedades por depósito lisosomal1,2. La presencia de consanguinidad o la existencia de más de un caso familiar de hidrops fetal de etiología desconocida, orienta la búsqueda hacia enfermedades metabólicas hereditarias.
Caso clínicoMujer de 35 años, de raza caucasiana y sin antecedentes familiares de interés. Entre los antecedentes personales destacan: episodios de ansiedad, cuadro de dolores articulares erráticos de etiología no filiada e intervención de hernia umbilical, grupo 0 Rh+. Como antecedentes obstétricos: un parto normal con una niña sana a término.
En la primera visita obstétrica realizada a las 13 semanas, con longitud craneocaudal (LCC) de 75,6mm y traslucencia nucal (TN) de 2,65mm (P90), el riesgo combinado para síndrome de Down resultó ser 1:2.212.
A las 17 semanas de gestación se realizó diagnóstico ecográfico de hidrops fetal, llevándose a cabo, tras una exploración exhaustiva de la anatomía fetal, las siguientes determinaciones en sangre materna: prueba de Coombs indirecto negativo, glucosa 6-fosfato-deshidrogenasa (G6PD) en rangos normales, Ac CMV IgG– IgM−, Ac. toxo IgG−, Ac. V herpes I+II IgG+ IgM−, RPR−, Ac. parvovirus B19 IgG+ IgM−. El cariotipo en líquido amniótico fue 46,XX.
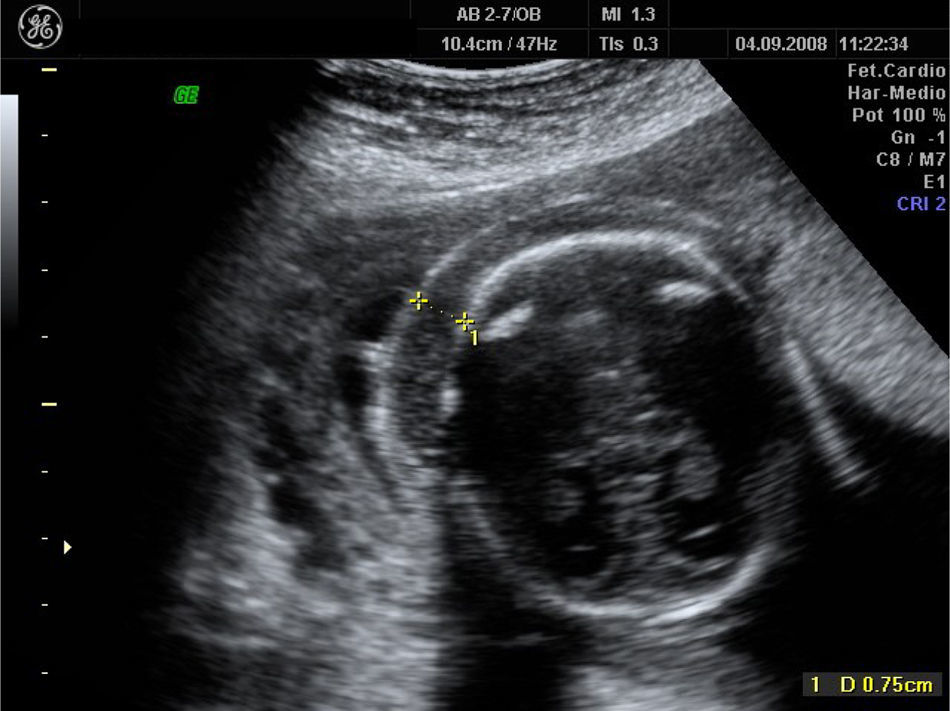
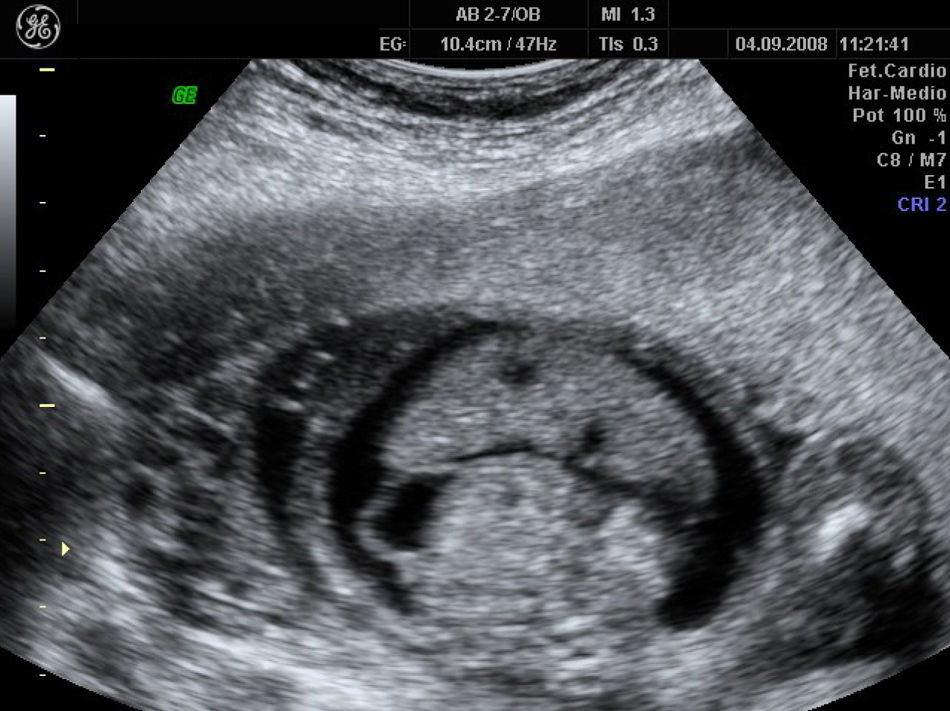
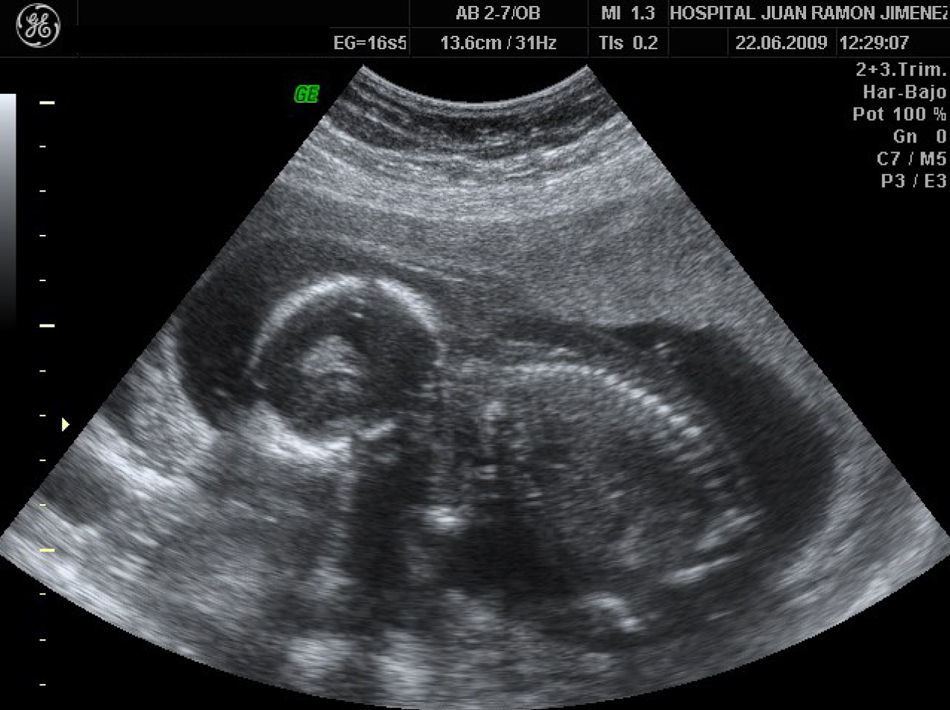
A las 20 semanas, con intensa ascitis y edema subcutáneo (figs. 1 y 2), la paciente solicitó interrupción legal por grave anomalía fetal.


El informe de la necropsia fetal indicó peso y talla bajas para la edad gestacional, sin malformaciones internas ni externas, evidenciándose a nivel de la cavidad abdominal abundante líquido libre claro.
Al año siguiente, la paciente consultó con una nueva gestación de 12+5 semanas, LCC de 65mm y TN de 1,71mm (P50), con riesgo combinado para síndrome de Down de 1:2.558.
A las 16+5 semanas de embarazo se detectó hidrops fetal, desplegándose de nuevo la batería diagnóstica: hemograma, bioquímica y estudio de coagulación normales, Coombs indirecto negativo, ANA-antiDNA-anticardiolipina negativos, serología de CMV y toxo negativas. Cariotipos paternos en sangre periférica normales. El cariotipo en líquido amniótico fue 46,XX.
Se analizó el líquido amniótico (sobrenadante y cultivo) siguiendo el protocolo habitual para descartar algunas de las enfermedades lisosomales que pueden causar hidrops fetal. Concretamente se analizaron los glucosaminoglucanos en el sobrenadante de líquido amniótico y se realizó el análisis enzimático en amniocitos cultivados de los enzimas beta-galactosidasa, beta-glucocerebrosidasa, beta-glucuronidasa y beta-hexosaminidasa. Con este protocolo se tiene la posibilidad de diagnosticar las mucopolisacaridosis (MPS) (excepto la IVA), la gangliosidosis GM1, la galactosialidosis, la enfermedad de Gaucher, la mucolipidosis II/III y la deficiencia múltiple de sulfatasas.
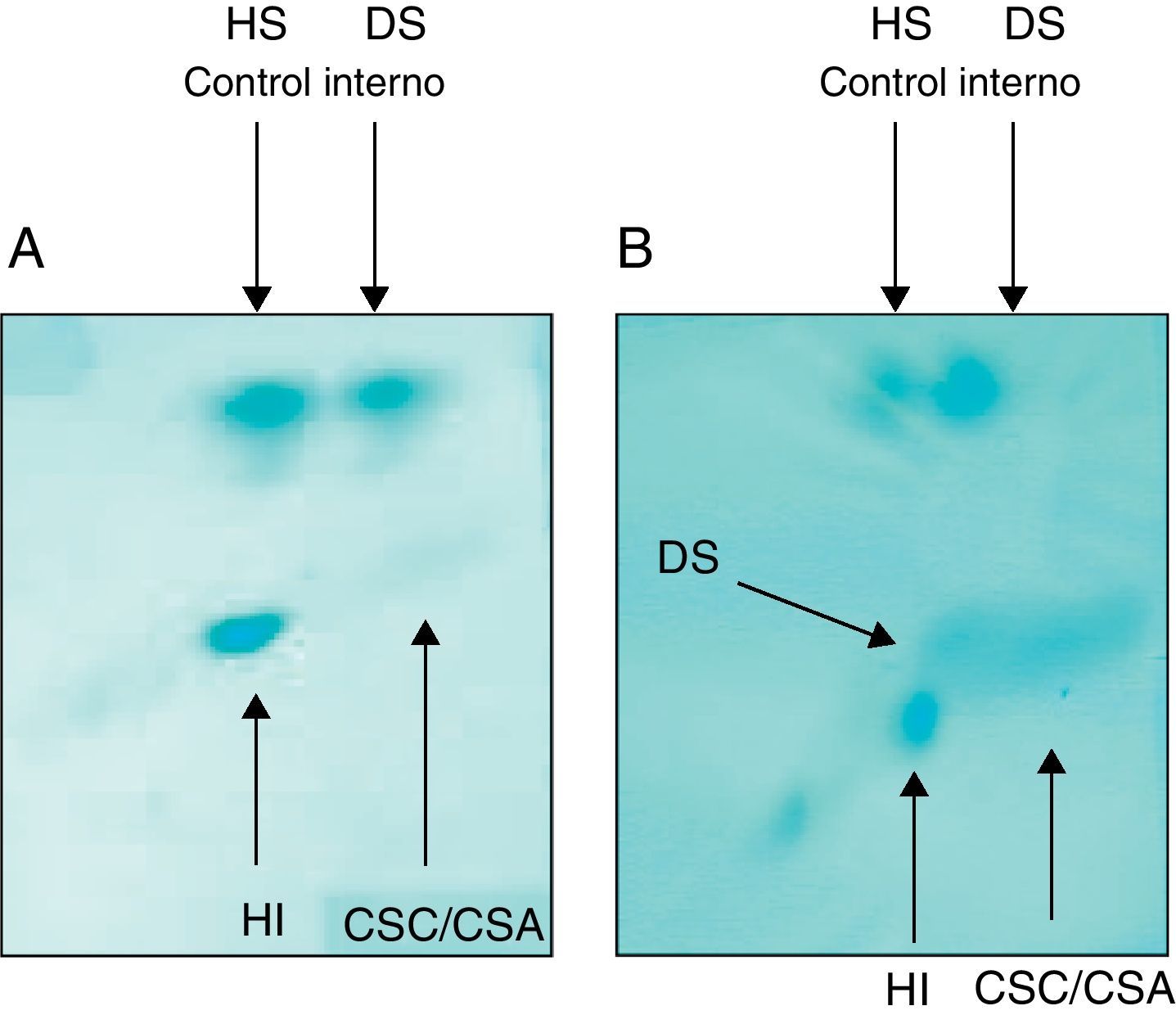
El feto presentó excreción elevada de condroitines sulfato A y C y dermatán sulfato en sobrenadante de líquido amniótico, indicativo de feto afecto de MPS tipos I, II o VII (fig. 3). La actividad beta-glucuronidasa fue de 2 nmol/h.mg (rango control: 89-247 nmol/h.mg). Este resultado confirmó que el feto estaba afecto de MPS tipo VII o enfermedad de Sly.
 Feto control. B) Feto con hidrops fetal, que presenta excreción de dermatán sulfato y condoitín sulfato A y C. CSA: condroitin sulfato A; CSC: condroitin sulfato C; DS: dermatan sulfato; HI: ácido hialurónico; HS: heparan sulfato.")
Electroforesis bidimensional en tiras de celulosa. A) Feto control. B) Feto con hidrops fetal, que presenta excreción de dermatán sulfato y condoitín sulfato A y C. CSA: condroitin sulfato A; CSC: condroitin sulfato C; DS: dermatan sulfato; HI: ácido hialurónico; HS: heparan sulfato.
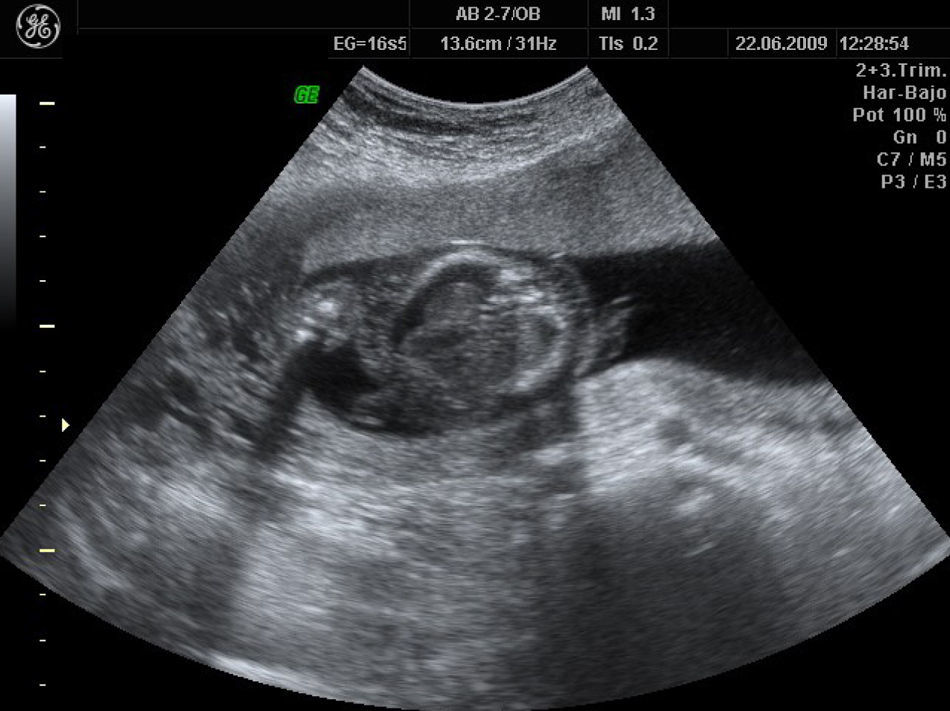
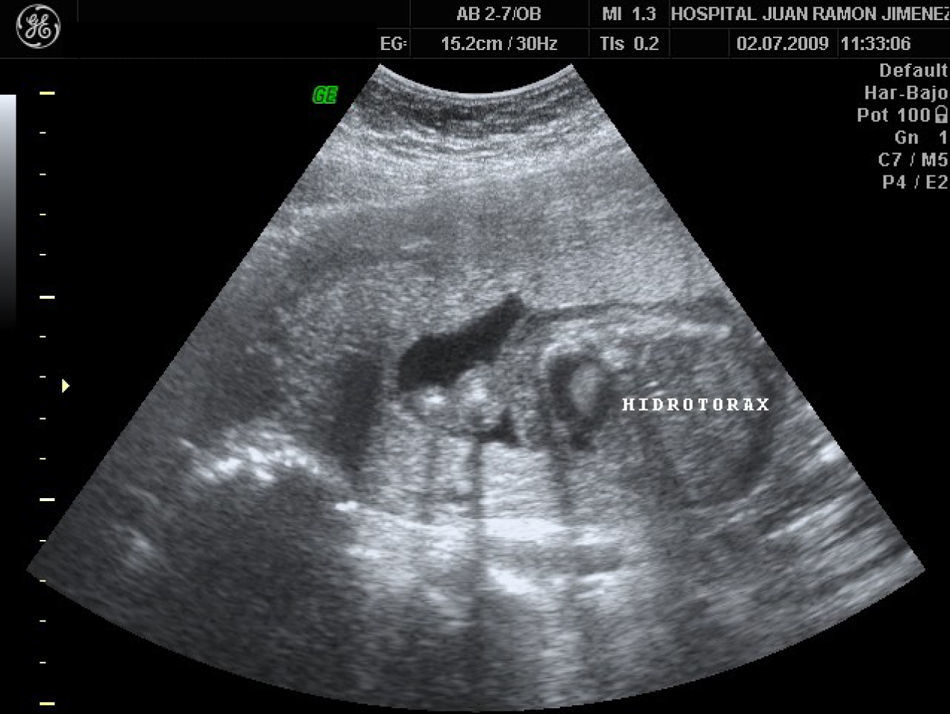
A las 18 semanas, con hidrotórax, ascitis y edema subcutáneo (figs. 4–6), la paciente decidió finalizar la gestación.
Discusión


Si bien el diagnóstico prenatal sonográfico de hidrops fetal es sencillo, el diagnóstico etiológico resulta complejo, no encontrando causa materna o fetal hasta en uno de cada 4 casos.
El diagnóstico exacto va a condicionar el manejo clínico y el consejo genético, por lo cual es fundamental realizar una exploración ecográfica sistematizada y minuciosa, intentando precisar la etiopatogenia. Además, se deben llevar a cabo otros exámenes maternos: hematológico, inmunohematológico, serológico y en líquido amniótico: inmunológicos, cromosómicos y/o bioquímicos3,4.
Distintos autores han publicado que las enfermedades metabólicas hereditarias y otros defectos monogénicos son responsables de entre el 1 y el 15% de todas las causas de hidrops fetal no inmune2,5–7, aunque las probabilidades de encontrar casos de hidrops fetal debido a enfermedades metabólicas hereditarias aumenta cuando se dan casos de recurrencia en la familia8–10, como sucede en el presente caso clínico.
Se han descrito más de 30 enfermedades metabólicas hereditarias causantes de hidrops fetal, entre ellas 15 enfermedades lisosomales11–13; con el protocolo utilizado se analizaron algunas de las más frecuentes: gangliosidosis GM1, galactosialidosis, enfermedad de Gaucher, MPS I, MPS VII, mucolipidosis II/III y deficiencia múltiple de sulfatasas. Las restantes enfermedades lisosomales, así como las otras enfermedades metabólicas hereditarias requieren una aproximación específica una por una y a veces no se puede completar el estudio en la etapa fetal, sino que se resuelve en el recién nacido; estas dificultades contribuyen a subestimar su incidencia, pero es importante perseverar en ello dado el riesgo de recurrencia familiar y la importancia de facilitar el oportuno asesoramiento genético.
En la MPS tipo VII, las manifestaciones clínicas pueden ir desde la muerte intraútero hasta formas más leves con supervivencia hasta la edad adulta: hidrocefalia, retraso mental, displasia esquelética, hepatoesplenomegalia crónica, dismorfia facial (puente nasal aplanado, hipertelorismo), entre otras14, aunque en el caso descrito, una presentación tan precoz del hidrops sugiere un pronóstico grave, probablemente incompatible con la vida.
La MPS VII no es susceptible de tratamiento fetal y el riesgo de recurrencia, teniendo en cuenta que su transmisión es autosómica recesiva, es del 25%.
En resumen, es de destacar la importancia del estudio de enfermedades metabólicas y, en concreto, de las enfermedades lisosomales, en los casos de fetos que presenten hidrops fetal no inmune, ya que su estudio podría permitir la identificación de la causa de esta afección, lo que resulta de gran utilidad de cara al consejo genético en la familia y para el diagnóstico prenatal de futuras gestaciones.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.