


Presentamos el caso de una mujer de 41 años, mexicana, con una vasculitis de vasos pequeños sistémica afectando riñón con una glomerulonefritis rápidamente progresiva pauciinmune y hemorragia pulmonar difusa. Una condición amenazante para la vida, que amerita un tratamiento intensivo inmediato con terapia combinada con metilprednisolona, ciclofosfamida y recambios plasmáticos. La paciente presentó mejoría transitoria, pero luego con deterioro progresivo y con hallazgos en la biopsia renal de mucha actividad, habiendo recibido tratamiento, se opta por terapia con fármacos depletadores de linfocitos B (rituximab) con el cual se obtuvo una mejoría sustancial del cuadro clínico e histológico, con remisión completa.
We report the case of a 41-year-old Mexican woman with systemic small vessel vasculitis affecting the kidney with pauci-immune rapidly progressive glomerulonephritis and diffuse pulmonary hemorrhage. This life-threatening condition warrants immediate intensive treatment with combination therapy with methylprednisolone, cyclophosphamide, and plasma exchange. The patient showed a transient improvement, with subsequent progressive deterioration. The results of renal biopsy showed high activity after treatment. B-cell lymphocyte depletion therapy (rituximab) was started, producing substantial clinical and histologic improvement, with complete remission.
La granulomatosis de Wegener es una vasculitis necrotizante de pequeños vasos, con inflamación granulomatosa involucrando tanto el tracto respiratorio superior e inferior como los riñones. El síndrome pulmón-riñón en la granulomatosis de Wegener, definida como una combinación de hemorragia pulmonar difusa y glomerulonefritis rápidamente progresiva, es una rara pero seria complicación con una alta tasa de mortalidad. Sin embargo, el tratamiento agresivo incluyendo inmunosupresión intensa es usualmente requerido para tratar la vasculitis subyacente.
Caso clínicoSe trata de mujer caucásica de 41 años de edad, dedicada a labores del hogar, procedente del Estado de México, sin antecedentes patológicos relevantes, con 3 meses de notar el aparecimiento de lesiones petequiales en extremidades inferiores. Se toman laboratorios sin hallazgos anormales, con creatinina (Cr) de 0,7mg/dl, siendo tratada con fármacos esteroides tópicos sin mejoría. Dos meses después, nota orina espumosa y edema leve de ambas piernas, acompañándose de disnea progresiva a máximos esfuerzos con tos seca en los últimos 15 días previos a nuestra admisión. Consulta con médico particular, quien envía laboratorios, encontrando Cr 4,9mg/dl, UN 59mg/dl por lo que es referida a nuestro instituto para valoración por Nefrología. No acude hasta 2 semanas después, en que nota reducción de volumen urinario y la disnea es mayor.
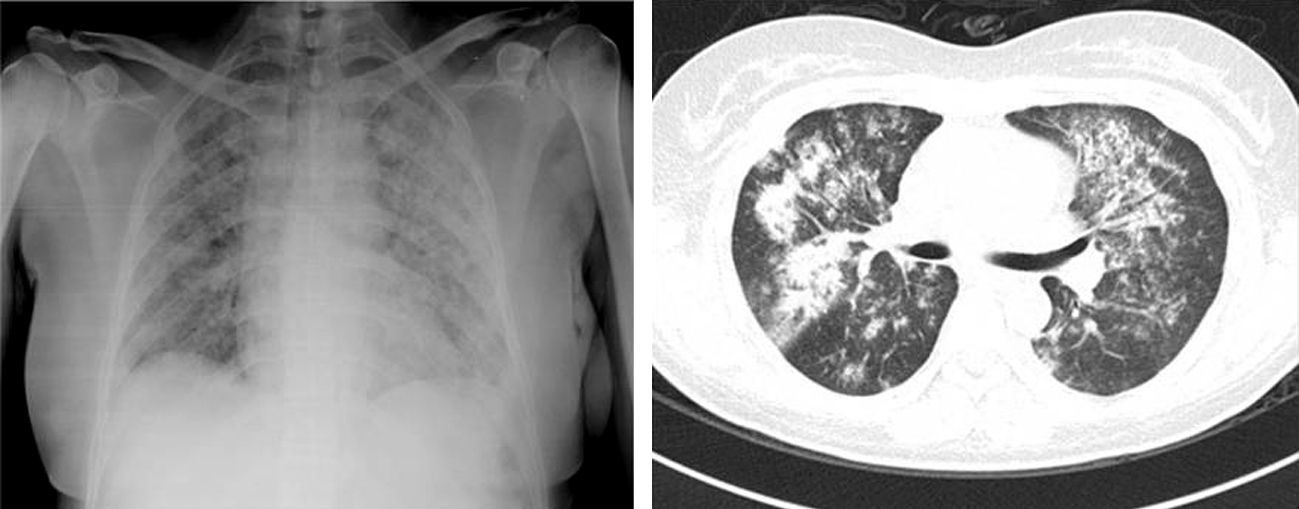
En la admisión, se recibe con marcado distrés respiratorio, tos con expectoración hemoptoica, con cianosis central. Su peso 62kg, talla 160cm, temperatura 36°C, saturando 80% a aire ambiente, con presión sanguínea 109/60mmHg, frecuencia cardíaca 129, frecuencia respiratoria 40 ciclos/min. Al examinarla, con intolerancia al decúbito dorsal, rinorrea mucopurulenta escasa, auscultándose estertores bilaterales teleinspiratorios difusos, extremidades con zonas hipercrómicas en pierna derecha, edema grado ii con fóvea. En sus exámenes a la admisión (tabla 1), se encontró leucocitosis con neutrofilia, sin eosinofilia, trombocitosis leve, con anemia severa, hiperazoemia importante, proteinuria subnefrótica (2.32g/24 h), con sedimento urinario activo y una gasometría arterial reportando hipoxemia severa. En la radiografía y la tomografía torácica se observaba imagen de vidrio despulido, con patrón reticular, acinar con tendencia a consolidar, de distribución difusa y engrosamiento de los septos interlobulillares (fig. 1).
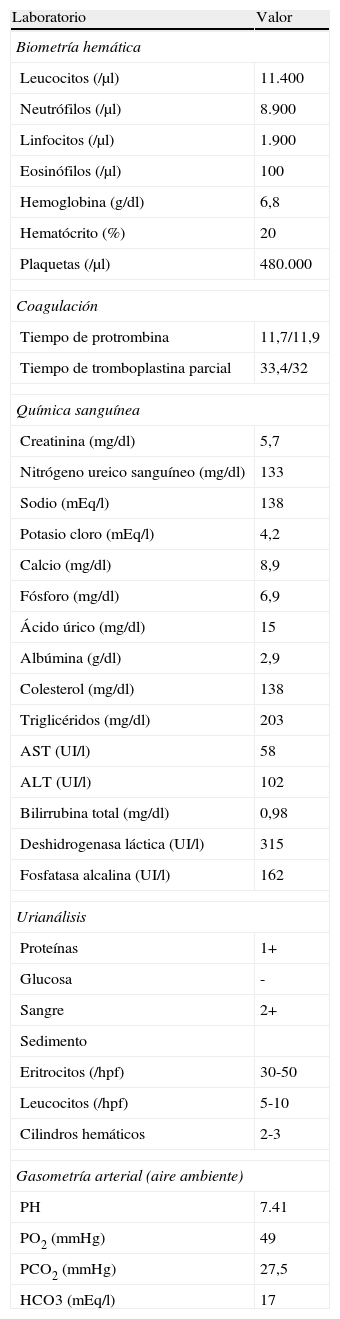
Laboratorios a la admisión
| Laboratorio | Valor |
| Biometría hemática | |
| Leucocitos (/μl) | 11.400 |
| Neutrófilos (/μl) | 8.900 |
| Linfocitos (/μl) | 1.900 |
| Eosinófilos (/μl) | 100 |
| Hemoglobina (g/dl) | 6,8 |
| Hematócrito (%) | 20 |
| Plaquetas (/μl) | 480.000 |
| Coagulación | |
| Tiempo de protrombina | 11,7/11,9 |
| Tiempo de tromboplastina parcial | 33,4/32 |
| Química sanguínea | |
| Creatinina (mg/dl) | 5,7 |
| Nitrógeno ureico sanguíneo (mg/dl) | 133 |
| Sodio (mEq/l) | 138 |
| Potasio cloro (mEq/l) | 4,2 |
| Calcio (mg/dl) | 8,9 |
| Fósforo (mg/dl) | 6,9 |
| Ácido úrico (mg/dl) | 15 |
| Albúmina (g/dl) | 2,9 |
| Colesterol (mg/dl) | 138 |
| Triglicéridos (mg/dl) | 203 |
| AST (UI/l) | 58 |
| ALT (UI/l) | 102 |
| Bilirrubina total (mg/dl) | 0,98 |
| Deshidrogenasa láctica (UI/l) | 315 |
| Fosfatasa alcalina (UI/l) | 162 |
| Urianálisis | |
| Proteínas | 1+ |
| Glucosa | - |
| Sangre | 2+ |
| Sedimento | |
| Eritrocitos (/hpf) | 30-50 |
| Leucocitos (/hpf) | 5-10 |
| Cilindros hemáticos | 2-3 |
| Gasometría arterial (aire ambiente) | |
| PH | 7.41 |
| PO2 (mmHg) | 49 |
| PCO2 (mmHg) | 27,5 |
| HCO3 (mEq/l) | 17 |

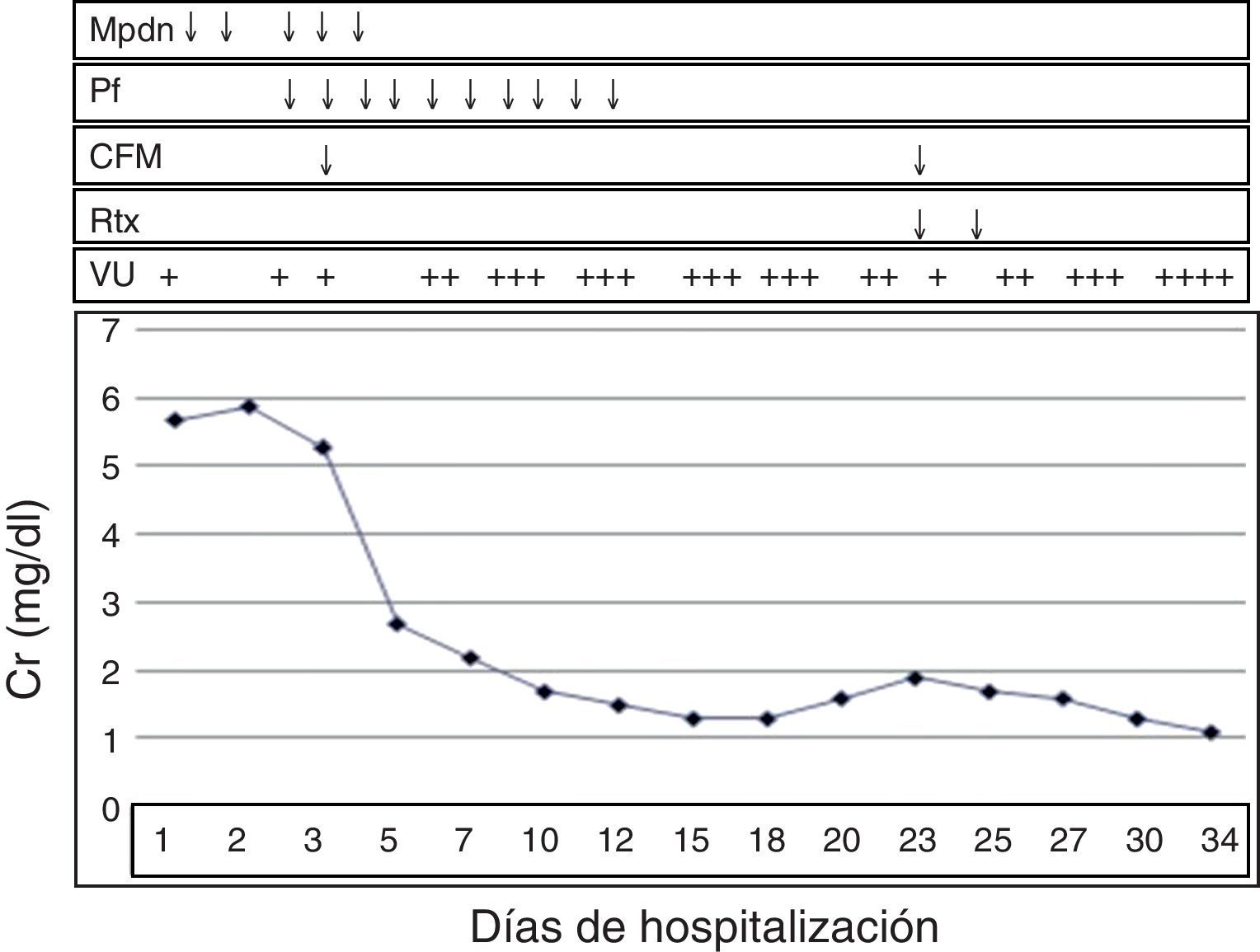
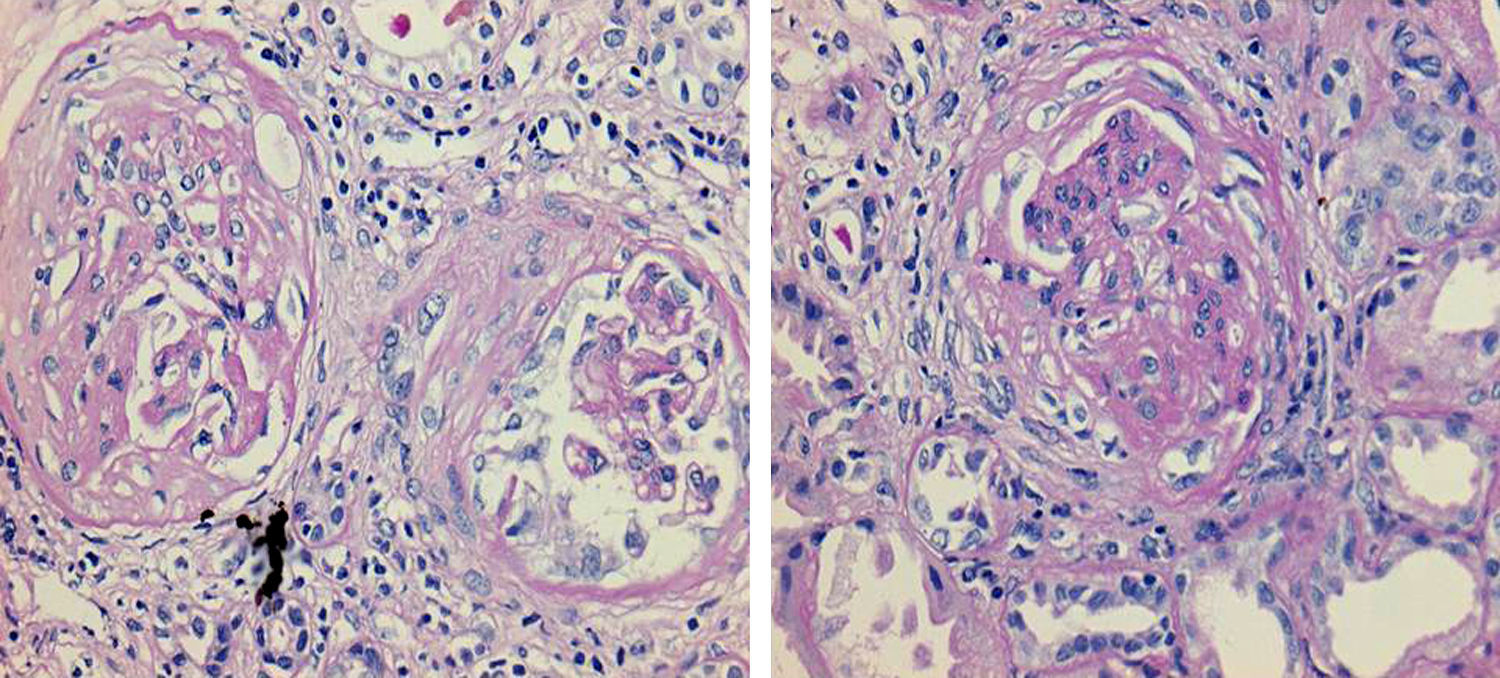
Debido a la presencia de hemoptisis, con infiltrados pulmonares bilaterales, anemia severa, marcadores inflamatorios elevados, antecedentes de lesiones purpúricas en extremidades, disfunción renal anúrica con comportamiento rápidamente progresivo y un sedimento urinario activo, se sospechó un síndrome pulmón-riñón secundario a vasculitis sistémica. Se inició de inmediato inmunosupresión con metilprednisolona (1.000mg/día) durante 5 días y sesiones de plasmaféresis diarias (# 10 sesiones). Se adicionó, además, un bolo de ciclofosfamida (1.000mg). Con este manejo intensivo tuvo mejoría clínica, con estabilización en la cifra de hemoglobina en 7,4g/dl sin requerir transfusión; hubo mejoría respiratoria a nivel pulmonar y en la función renal, con descenso de Cr hasta 1,9mg/dl y recuperación de volumen urinario progresivo hasta 1.500ml/24 h (fig. 2). Todos los cultivos se reportaron negativos. Se decidió realizar biopsia renal percutánea al día 18 de hospitalización, cuyo reporte histopatológico fue compatible con esclerosis global (15/29 glomérulos) y 4 más en vías de esclerosis con semilunas fibrosas. En los analizables, se encontró proliferación extracapilar fibrocelular con abundantes células inflamatorias y cariorrexis, la tercera parte de ellos con necrosis fibrinoide segmentaria y esclerosis segmentaria cicatricial en 4 glomérulos y fibrosis intersticial del 25% (fig. 3). La inmunofluorescencia directa resultó negativa para todos los inmunorreactantes. Estos hallazgos fueron descritos como glomerulonefritis proliferativa extracapilar con lesiones necrosantes y esclerosantes segmentarias compatibles con vasculitis pauciinmune.

Evolución clínica del caso. Curva creatinina/tiempo durante hospitalización. La pasmaféresis y corticosteroides mejoraron función renal pero el rituximab permitió una remisión del cuadro clínico. CFM: ciclofosfamida; Mdpn: metilprednisolona endovenoso; Pf: plasmaféresis; Rtx: rituximab; VU: volumen urinario.
.")
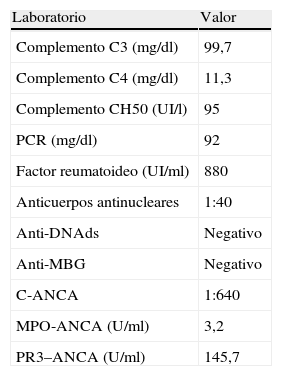
Al día 2 de su hospitalización se midieron anticuerpos anticitoplásmicos de neutrófilos (ANCA), resultando positivos para proteinasa 3 (PR3) con valor de 145,7 U/ml, negativos para mieloperoxidasa (MPO) con valor de 3,2 U/ml (tabla 2), por lo que, considerando la ausencia de antecedente de asma y eosinofilia, la presentación clínica, los hallazgos histológicos de vasculitis de pequeños vasos y una óptima respuesta a inmunosupresión, se catalogó como una granulomatosis de Wegener, aun cuando no fue posible demostrar una inflamación granulomatosa.
Test serológicos a la admisión
| Laboratorio | Valor |
| Complemento C3 (mg/dl) | 99,7 |
| Complemento C4 (mg/dl) | 11,3 |
| Complemento CH50 (UI/l) | 95 |
| PCR (mg/dl) | 92 |
| Factor reumatoideo (UI/ml) | 880 |
| Anticuerpos antinucleares | 1:40 |
| Anti-DNAds | Negativo |
| Anti-MBG | Negativo |
| C-ANCA | 1:640 |
| MPO-ANCA (U/ml) | 3,2 |
| PR3–ANCA (U/ml) | 145,7 |
En su evolución clínica, luego de la metilprednisolona, continuó con prednisona a 1mg/kg/día, pero inició con ascenso progresivo de Cr y reducción nuevamente de volumen urinario al día 23 de su hospitalización, por lo que, considerando los hallazgos de lesiones extracapilares activas encontradas en la biopsia renal, luego de recibir inducción a una inmunosupresión con esteroides, ciclofosfamida y plasmaféresis, se consideró la posibilidad de «resistencia» a inmunosupresión convencional, por lo que se decidió administrar rituximab en 2 dosis (500mg/dosis) con intervalo de 7 días. Posterior a lo cual descendió la Cr hasta 1,1mg/dl, mejoró el volumen urinario de 2.000ml/24h, la proteinuria de 300mg/24 h, manteniéndose estable y pudo ser egresada. En su seguimiento, a un mes de su hospitalización con títulos negativos de P-ANCA de 6,2 UI/ml y C-ANCA 2,9 UI/ml, se realiza una segunda biopsia renal, encontrando 6/15 glomérulos con esclerosis global, en 4 de ellos con semilunas fibrosas, 4/15 glomérulos con esclerosis segmentaria, el resto normales y sin hallazgos de actividad. Se continuó con bolos mensuales de ciclofosfamida y prednisona 0,5mg/kg/día, hasta la fecha actual con esteroides en descenso a 4 meses del evento, persiste con títulos negativos de ANCA, Cr 1,2mg/dl y proteinuria 220mg/24 h.
DiscusiónPresentamos el caso de una paciente con un síndrome de pulmón–riñón como complicación de una vasculitis de vasos pequeños asociada a ANCA, catalogada como granulomatosis de Wegener al carecer de antecedentes de asma, eosinofilia y cursar con involucro del tracto respiratorio superior bajo la forma de rinitis (síntomas E), pulmones con hemorragia alveolar difusa (síntomasL), deterioro rápidamente progresivo de la función renal con hematuria/proteinuria y edemas (síntomasK), glomerulonefritis crescéntica necrotizante sin depósito de complejos inmunes (criterio histológico) y positividad para PR3-ANCA/C-ANCA, según los criterios propuestos por Research Group of Intractable Vasculitis, Ministry of Health, Labor, and Welfare (MHLW) de Japón, país con una alta prevalencia e incidencia de la granulomatosis de Wegener1,2.
Si bien la meta debe ser no sobretratar la enfermedad leve y no infratratar la enfermedad severa, el manejo que se le brindó fue el descrito por la literatura en la inducción a la inmunosupresión con glucocorticoides y ciclofosfamida, considerando que los corticosteroides solos no son tan efectivos para la terapia de inducción como cuando se combinan con ciclofosfamida con un 75% de remisión a los 3 meses y un 90% a los 6 meses, adicionando la plasmaféresis por la severidad de su presentación y el alto riesgo de muerte que le confería la hemorragia pulmonar3. Pocos estudios puntualizan en el uso de la plasmaféresis para tratar el síndrome pulmón-riñón de la vasculitis de vasos pequeños asociada a ANCA. Gallagher et al. trataron a 12 de 14 pacientes con síndrome pulmón-riñón con un promedio de 6,1 plasmaféresis adicionales a corticoesteroides y ciclofosfamida iniciada de manera temprana, con un seguimiento de 22±9 meses. Tuvieron una sobrevida de 85% a un año y 67% a 2 años, si bien 2 de estos pacientes tenían anticuerpos antimembrana basal asociados (anti-MBG). Se ha descrito que pacientes con vasculitis de pequeños vasos asociada a ANCA que tengan de forma simultánea enfermedad anti-MBG o enfermedad por complejos inmunes deben ser tratados similarmente a pacientes con enfermedad ANCA aislada3,4. Klemmer et al. han reportado 20 pacientes con vasculitis ANCA asociada y hemorragia pulmonar difusa, tratados con un promedio de 6,4 recambios plasmáticos, con resolución de la hemorragia pulmonar en el 100% de los casos, 50% de ellos con mejoría en la función renal, concluyendo que el inicio temprano de la plasmaféresis junto a terapia inmunosupresora agresiva es salvadora del componente pulmonar del síndrome5. El papel del recambio plasmático como un componente de la terapia de inducción se ha definido de utilidad en 2 poblaciones específicas: 1) hemorragia pulmonar amenazante de la vida y 2) pacientes con falla renal dependiente de diálisis al tiempo de la presentación. Se ha demostrado su utilidad en la reducción de la mortalidad asociada a hemorragia pulmonar en comparación con los controles históricos5. De igual forma, hay estudios que sugieren que los recambios plasmáticos comparados con metilprednisolona incrementan la tasa de recuperación de la falla renal, atribuyéndoles a estos un 24% de reducción del riesgo de progresión a enfermedad renal crónica terminal en comparación con los corticosteroides (43 a 19% a un año)6. Otros autores como Walsh et al., en su metaanálisis sobre la utilidad de la plasmaféresis en vasculitis de vasos pequeños para pacientes con enfermedad renal severa, con las limitantes estadísticas descritas por los autores, han concluido que no existe evidencia de que reduzca el riesgo de muerte, sí existe reducción en la mortalidad asociada a falla renal terminal y hay poca evidencia de que reduzca el riesgo de falla renal como monoterapia7. En el caso de nuestra paciente, se realizaron 10 recambios plasmáticos. Luego, hubo necesidad de ser más agresivos en el tratamiento empleado dada la evidencia histológica de hallazgos muy activos a nivel renal, así como el deterioro en la función renal evidente al día 23 de su evolución, considerando que, previamente a la biopsia renal, había sido tratada. Se decidió emplear rituximab al no obtener resultados satisfactorios con corticosteroides y ciclofosfamida.
El empleo de terapias novedosas biológicas como el rituximab en las vasculitis de vasos pequeños ha sido descrito en casos de sospecha de «resistencia a ciclofosfamida» la cual es rara, pero puede ser definida como la presencia de enfermedad activa afectando un órgano mayor a pesar de la dosis óptima de ciclofosfamida y corticosteroides1. Al respecto, ha sido sugerido que la eliminación de linfocitos B con anticuerpos anti-CD20 (rituximab) podría tener un efecto favorable en la granulomatosis de Wegener por remoción de células responsables de producir los ANCA. El rituximab y la alta dosis de glucocorticoides parece ser de beneficio. Keogh et al. reportan, en un estudio de 11 pacientes con vasculitis de vasos pequeños refractaria, una remisión completa alcanzada en todos los pacientes. Todos los pacientes tenían enfermedad refractaria definida como enfermedad activa persistente a pesar de máxima dosis tolerada de ciclofosfamida o una contraindicación para su administración, siendo tratados con 4 dosis de 375mg/m2 además de prednisona a 1mg/kg/día8. Existen al menos 10 series de casos reportando su uso con resultados promisorios al mantener a los pacientes en remisión completa y depletar los títulos de ANCA9. Se ha descrito una tasa de remisión completa en 2 a 6 meses, alrededor del 75% usando rituximab, con una depleción de linfocitos B periféricos a través de determinación de CD19 y negativización de los ANCA; sin embargo, se espera una recuperación de los linfocitos a los 10 meses, lo que constituye un riesgo de recaída, por lo que se ha descrito su uso en dosis repetidas de forma preventiva10. Sin embargo, deben considerarse los riesgos a mediano y largo plazo por el uso de estos fármacos, lo que obliga a un seguimiento estrecho de los pacientes. Aunque la combinación de rituximab+corticoesteroides ya fue aprobada por la FDA como terapia de primera línea para inducción a la remisión en granulomatosis de Wegener y poliangiitis microscópica, las cuestiones relacionadas con la terapia de mantenimiento después del uso de rituximab y su toxicidad con el uso simultáneo de ciclofosfamida siguen sin respuesta, requiriendo para ello estudios controlados aleatorizados en el futuro11.