



Three leaders of computational chemistry received this year the Nobel Prize in Chemistry. Molecular Mechanics was the discipline they created to account for the formation and breaking of bonds, what is fundamental for chemistry. Michael Levitt, Arieh Warshel and Martin Karplus combined quantum mechanics and molecular mechanics and they have received the Nobel Prize in Stockholm last December 10th.
Los líderes de la química computacional fueron galardonados este año con el Premio Nobel de Química. La mecánica molecular funciona y permite estudiar interesantes problemas químicos. Pero a esta técnica le hacía falta tomar en cuenta la posibilidad de la formación y el rompimiento de enlaces químicos, es decir, le faltaba incluir la reacción química misma. Y es la resolución de esta carencia fundamental, en particular, lo que premió el comité Nobel. Los tres galardonados (Michael Levitt, Arieh Warshel y Martin Karplus) contribuyeron en la combinación de dos metodologías —mecánica cuántica y mecánica molecular— para tratar sistemas complejos y lo recibieron el pasado 10 de diciembre en Estocolmo.
El premio Nobel de Química se otorga ya sea por el descubrimiento de algún fenómeno químico excepcional —necesariamente experimental— o por el desarrollo de una técnica que haya permitido el descubrimiento, la invención o la justificación de fenómenos químicos excepcionales, lo que permite que esta técnica pueda ser experimental o teórica. La lista de las contribuciones teóricas que han recibido el premio en los últimos sesenta años es relativamente escasa [NobelPrizeLaureates]:
- •
Linus Pauling, 1954, “por su investigación en la naturaleza del enlace químico y su aplicación a la elucidación de la estructura de sustancias complejas”.
- •
Robert Mulliken, 1966, “por su trabajo fundamental concerniente a los enlaces químicos y la estructura electrónica de moléculas mediante el método del orbital molecular”
- •
Kenichi Fukui y Roald Hoffman, 1981, “por sus teorías concernientes al curso de las reacciones químicas”
- •
Rudolph Markus, 1992, “por sus contribuciones a la teoría de las reacciones de transferencia de electrones en sistemas químicos”
y más recientemente, cuando parte de la química teórica se empezó a llamar química computacional,
- •
Walter Kohn y John Pople, 1998, “por su desarrollo de la teoría de funcionales de la densidad” y “por su desarrollo de los métodos computacionales de la química cuántica”, respectivamente
para llegar al que nos ocupa en este año
- •
Martin Karplus, Michael Levitt y Arieh Warshel, 2013, “por el desarrollo de modelos multiescala para sistemas complejos”
Estos dos últimos premios representan la confirmación de que la química cuenta con una nueva técnica general, la química computacional. En palabras de un atrevido reportero de The Economist, “En la vida real, esto podría significar que eventualmente la mayoría de los experimentos químicos se van a realizar en el silicio de los chips en vez de en el material de vidrio de los laboratorios”. No hemos llegado a ese punto, pero nos acercamos poco a poco con proyectos como la Iniciativa del Genoma de los Materiales, que promueve el Gobierno de Estados Unidos [MaterialsGenome] y Folding at Home para resolver el complejo problema del plegamiento de proteínas [Lane et al. 2013].
Dos cosas han permitido la entronización de la química computacional como una nueva herramienta general de la química. La primera es la que también ha definido nuestras vidas en la actualidad: la capacidad en hardware y software de nuestras computadoras. Considérese que los detalles fundamentales de la ciencia que fundamenta la realización de química in silico se conocen desde hace cuando menos setenta años, y lo que ha permitido su aplicación es el aumento en las capacidades computacionales de las que disponemos —un incremento de un millón de veces en los últimos cuarenta años. Claro que por este aumento computacional no se dan Nobeles de Química. Es la segunda razón de la entronización de la química computacional la que ha merecido el Nobel en 1998 y el de este año: el desarrollo de las técnicas físicoquímicomatemáticocomputacionales que han garantizado el gran éxito en la resolución de problemas químicos por estos métodos.

La mecánica cuántica es la teoría que se debe usar para estudiar el enlace químico y su modificación, es decir el proceso de reacción química. Sin embargo, su aplicación es increíblemente compleja e, incluso luego de hacer arriesgadas y valientes aproximaciones —como las que hicieron merecedores a Pople y Kohn del Nobel en 1998— el tamaño de los sistemas químicos que se pueden tratar es relativamente pequeño: mil electrones o por ahí es el límite presente con las computadoras más avanzadas. Pero ésta es una grave limitación. Porque no es solo que la química sea compleja y que en muchas de sus aplicaciones más fundamentales haga falta tomar en cuenta muchos más que mil electrones, sino que algunas de sus aplicaciones más importantes —por ejemplo, las relacionadas con la bioquímica— requieren el estudio de sistemas que tienen miles de veces esa cantidad de electrones.
Y aquí es donde inicia esa subdisciplina de la química computacional cuyos líderes fueron galardonados este año. La mecánica molecular plantea una alternativa al problema del tamaño en el estudio de la química reconociendo que muchos fenómenos químicos de importancia no incluyen la creación/formación de enlaces, sino que son regidos por la interacción entre moléculas que conservan su estructura primaria original a lo largo del proceso. Estas moléculas interactúan de manera tal que su conformación —estructura secundaria, terciaria, cuaternaria— se modifica y, al hacerlo, define el fenómeno que se observa; pero su esencia, determinada por el enlace químico se mantiene intacta. Ésta es, entonces, la mecánica de las moléculas.
Bajo este paradigma no hace falta tratar cuánticamente a las moléculas. Las interacciones que definirán su conformación —y por tanto el fenómeno químico deseado— pueden tratarse clásicamente. Y si bien el tratamiento clásico no es de ninguna manera nomás enchílame otra, sí es considerablemente más sencillo que el tratamiento cuántico, de tal forma que se puede ampliar importantemente, por un lado, el tamaño de los sistemas a considerar —y pasar del máximo de 20 aminoácidos promedio que se pueden tratar cuánticamente con mil electrones— a, por ejemplo, los 2,600 aminoácidos, más las 90,000 moléculas de agua, considerados por Domínguez y colaboradores recientemente Domínguez et al. 2013; y se puede también, por otro lado, incluir otros fenómenos fundamentales para el estudio químico del proceso, como por ejemplo, el efecto de la temperatura o la dependencia temporal. La mecánica molecular permite, entonces, aumentar mil veces el tamaño de los sistemas considerados e incluir variables adicionales en el estudio de los procesos químicos.
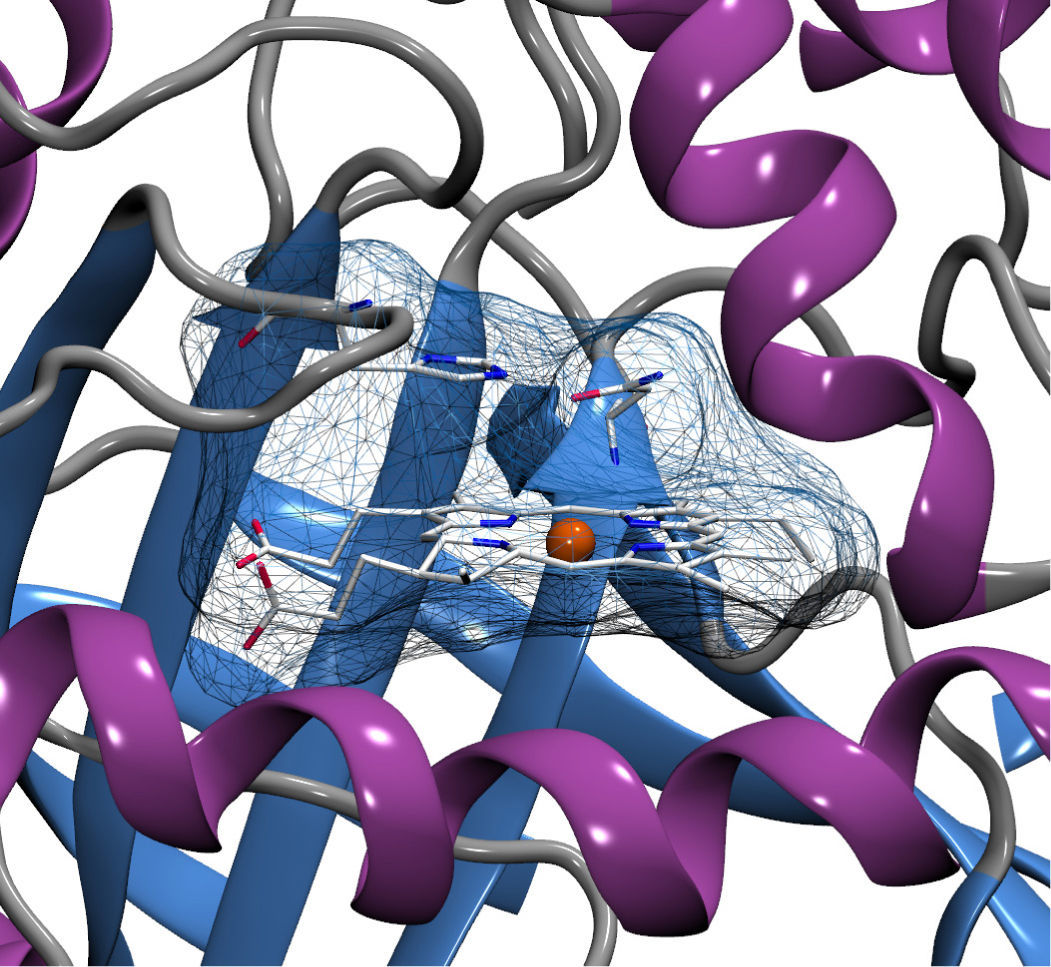
Mecánica molecularEn la mecánica molecular se considera a una molécula como una colección de masas centradas en el núcleo de los átomos y que están conectadas entre sí por resortes (enlaces) que se estiran, desplazan y rotan —sin romperse ni crearse— como resultado de aplicarles un potencial.
La mecánica molecular considera que un conjunto de fuerzas físicas pueden ser usadas para describir geometrías y energías moleculares. Entonces, el espacio conformacional que se obtiene es un ajuste de geometrías que busca minimizar la energía interna de las moléculas. La mecánica molecular involucra la construcción de un potencial de energía aplicado a un conjunto de posiciones atómicas —extraídas experimentalmente— para calcular un espacio de conformaciones o simulaciones de dinámica molecular que podrá explicar los estados energéticamente accesibles de un sistema con respecto al tiempo.
De acuerdo con la mecánica clásica la descripción de la energía interna de las moléculas consiste en la suma de la energía cinética, K, y potencial, V,
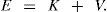
La energía cinética consiste en la suma del movimiento de todos los átomos del sistema —controlando, por ejemplo, la temperatura del sistema—; la energía potencial consiste en la suma de las interacciones entre los átomos y se divide en dos contribuciones: las que provienen de la interacción de enlace definida por las uniones covalentes y que mantienen a dos o más átomos juntos y la contribución que viene de las interacciones de no enlace.
El conjunto de estas interacciones se denomina campo de fuerzas. La interacción de enlace incluye la modificación de la energía de enlace debida al cambio de la distancia entre los átomos —típicamente en la aproximación armónica—, la modificación de la energía debida al cambio en el ángulo entre cada tres átomos enlazados —que se trata también como un potencial armónico— y las rotaciones entre cada grupo de cuatro átomos que definen un ángulo diedro —que se representa como una función periódica. Por otro lado, las interacciones de no enlace se dividen en dos: interacciones electrostáticas que se describen con la ley de Coulomb e interacciones atractivas y repulsivas descritas con un potencial de LennardJones.
Se han desarrollado diferentes campos de fuerza para describir biomoléculas, cada uno de ellos con ventajas particulares; charmm, uno de los campos de fuerza más populares fue desarrollado en Harvard por Martin Karplus. En este campo de fuerza, todos los átomos se toman en cuenta o, como se dice en la jerga de mecánica molecular, todo los átomos son explícitos. En cambio existen otros campos de fuerza como gromos, desarrollado por Wilfred van Gunsteren, el cual propone un paso más en la idealización del sistema al definir conjuntos de átomos unidos.
El premio Nobel de Química 2013La mecánica molecular funciona y permite estudiar interesantes problemas químicos. Pero a esta técnica le hacía falta tomar en cuenta la posibilidad de la formación y el rompimiento de enlaces químicos, es decir, le faltaba incluir la reac ción química misma. Y es la resolución de esta carencia fundamental, en particular, lo que premió el comité Nobel. Los tres galardonados contribuyeron en la combinación de las dos metodologías —mecánica cuántica y mecánica molecular— para tratar sistemas complejos: usaron mecánica clásica para describir un sistema con muchos átomos que sería imposible abordar con pura química cuántica y mecánica cuántica para estudiar la región del sistema que lleva a cabo una reacción química.
El desarrollo conjunto de la metodología de multiescala comenzó cuando Arieh Warshel y Martin Karplus se juntaron a principios de la década de los setenta. Warshel era experto en los potenciales intermoleculares descritos por la mecánica clásica y Karplus tenía una importante experiencia en química cuántica. Su idea era juntarse para aplicar ambas metodologías y calcular la estructura atómica de un cromóforo involucrado en la visión animal. En un trabajo conjunto, calcularon los núcleos de los átomos usando una aproximación clásica y los electrones fueron modelados usando química cuántica. Pocos años después Arieh Warshel y Michael Levitt pudieron describir exitosamente la frontera entre la parte del sistema estudiado mediante mecánica clásica de la parte electrónica descrita con química cuántica.
Todavía en un segundo paso, Michael Levitt y Arieh Warshel con la idea de estudiar sistemas aún más grandes y/o procesos que se llevan a cabo en tiempos muy largos —como el plegamiento de proteínas— iniciaron el desarrollo de una aproximación más: agruparon conjuntos de átomos en unidades rígidas para tratarlos como seudoátomos clásicos; idea que habría de evolucionar en la creación de los ahora llamados modelos de grano grueso.
Con la química computacional así combinada se ha podido caracterizar con detalle la estructura, el movimiento y la función biológica de biomoléculas. Por ello, los fundadores de la metodología de multiescala para sistemas químicos complejos han sido galardonados con el premio Nobel de Química que ha sido entregado en diciembre de 2013.
Los chismesEl chisme no es ajeno al premio Nobel. Más bien es una de sus características peculiares. Desde el que cuenta de multitud de profesores que secuestran el teléfono de su oficina al empezar octubre para que nadie ose interponerse ante la potencial llamada de Estocolmo, hasta los comentarios de muchísimos químicos profesionales acerca de la relevancia y justicia del tema premiado y de las personas premiadas. Porque en los tiempos actuales una disciplina tiene siempre muchos líderes y siempre cabrá la duda de si la selección de los tres premiados no dejó fuera a un posible cuarto —o alternativa para el tercero— con más merecimientos.
Sin embargo, en este año destaca el comentario de algunos de los científicos de la disciplina más cercana posible a la premiada: los químicos cuánticos.
Académico 1: Ésta es la mejor botella que tengo, me gustaría regalársela a mi colega que acaba de ganar el Nobel de Química. Académico 2: ¿No crees que le deberías amarrar una Budweiser a la caja? Porque básicamente eso es la QM/MM. Académico 3: Amárrale una Bud Light.
Martin Karplus es, como muchos de los profesores de Harvard, respetuosamente temido y temerosamente respetado por la comunidad harvardiana. No cualquiera se anima a entablar conversación con él. Nos llamó la atención, por tanto, la cándida autobiografía que escribió hace algunos años. Vale la pena leerla Karplus, 2006.
Académico 4: Lo bueno es que ahora sí vamos a poder trabajar tranquilos porque ya no hay que pelear por el Nobel para un teórico. Seguramente van a pasar muchos años antes de que se otorgue otro a esta disciplina.
No compartimos la opinión de algunos de los colegas anteriormente mencionados. Quizás el periodista de The Economist tenga razón y en el futuro cercano, con base en las técnicas de la química computacional y apoyados como siempre por el increíble avance de las capacidades computacionales y el análisis de datos —eso que se llama ahora Big Data— otro teórico reciba un premio Nobel por el descubrimiento, in silico, de algún fenómeno químico excepcional.