



It is common that lectures and manuscripts in Biochemistry deal with the Levinthal padarox. Levinthal wondered how long it would take for a protein to fold correctly if it had to sample all possible conformations in three-dimensional space. Small proteins usually fold spontaneously within seconds and even the largest proteins fold within minutes. However, the Levinthal's calculations show that any protein needs an infinite time to fold correctly. This problem is basic in life and here we review some of the most important contributions to try to resolve this paradox.
Una paradoja es una idea extraña o irracional que se opone al sentido común y a la opinión general. En pocas palabras, una paradoja es una idea que confronta lo que la lógica permite concluir, aunque posee una serie de factores que se consideran válidos o reales. La mayoría de las veces, la adquisición de nuevo conocimiento provoca que dicha paradoja tome sentido o se resuelva. Así, las paradojas han impulsado importantes avances en la ciencia y filosofía. Lo que es imprescindible en estos casos es un correcto uso de las capacidades de abstracción de la mente para lograr una adecuada comprensión de cualquier paradoja. Por lo tanto, su objetivo no es lograr que el individuo contribuya con ideas imaginativas y fabulosas para su resolución. Sin embargo, en caso de lograrlo, las aportaciones a ciertas áreas del conocimiento pueden ser relevantes.
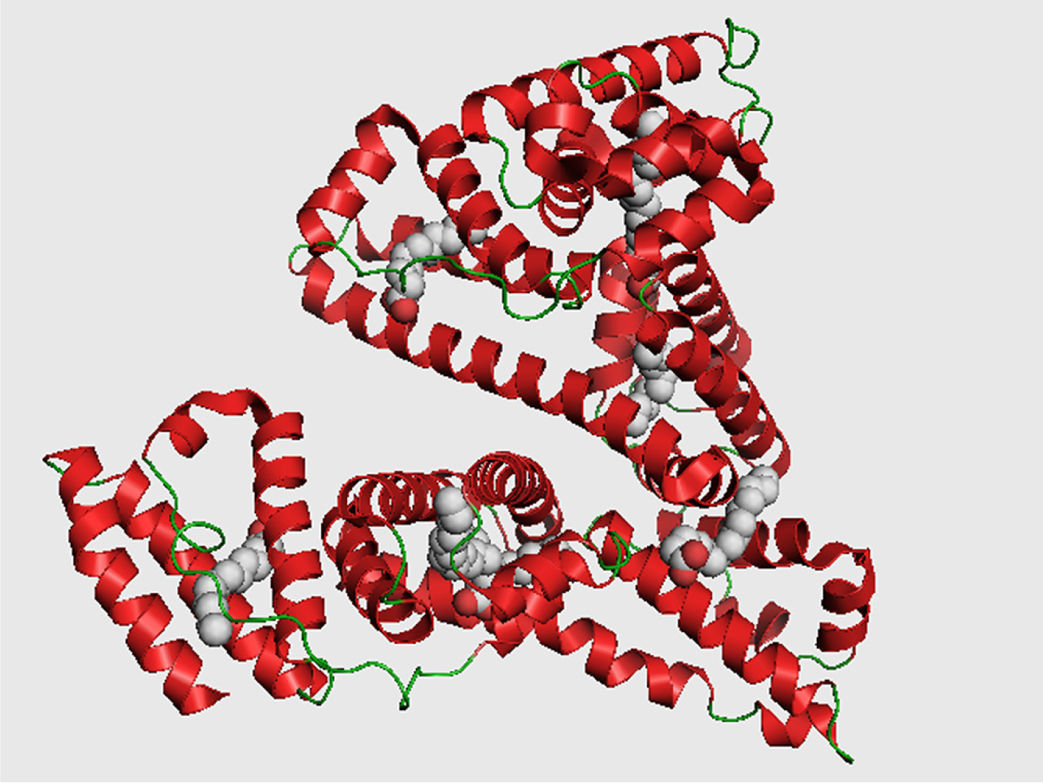
Quizás una de las paradojas más excitantes es la planteada en 1969 por Cyrus Levinthal (Levinthal, 1969), en ese tiempo profesor del Departamento de Ciencias Biológicas de la Universidad de Columbia. Imagine una proteína, moléculas formadas por una cadena larga y flexible de aminoácidos como la mostrada en la figura 1. Su función está ligada íntimamente a la distribución espacial que adopta durante su plegamiento. Sin embargo, si una proteína se plegara de forma aleatoria, muestreando el espacio conformacional de manera exhaustiva, entonces la localización de la estructura tridimensional correcta le tomaría una cantidad enorme de tiempo, aun cuando cada una sus conformaciones sean adoptadas y su función evaluada a una velocidad extremadamente rápida (en escalas de tiempo del orden de nano y picosegundos). En otras palabras, si suponemos que cada enlace peptídico tiene sólo tres grados de libertad, entonces para una proteína pequeña de unos 101 aminoácidos existirán 3100 = 5 × 1047 conformaciones. Aun si la proteína fuera capaz de explorar estas conformaciones a una enorme velocidad, digamos de 1013 conformaciones por segundo, lo que significa 3 × 1020 por año, entonces le tomaría 1027 años probar todas las posibilidades, es decir, si la edad del Universo se estima en 1.37 × 1010 años —13 mil 700 millones de años—, una proteína requeriría un tiempo de 17 órdenes de magnitud mayor que la edad del Universo para plegarse! Por lo tanto, empleando esta lógica parece imposible que las proteínas encuentren su estructura óptima, a pesar de que el tamaño promedio de las cadenas polipeptídicas oscila alrededor de los 300 aminoácidos. Sin embargo, una proteína se pliega en una escala de tiempo ¡de segundos o menos!
 y su masa molecular es de 67 kdaltons.")
Levinthal propuso que la búsqueda conformacional estocástica no es el mecanismo real del plegamiento, sino que éste debe ser un proceso dirigido por las condiciones fisicoquímicas existentes durante la biosíntesis de la proteína y las rutas bioquímicas que experimentan a posteriori, o sea una vez plegadas (Levinthal, 1968 y 1969).
Para entender mejor la idea de Levinthal necesitamos introducir brevemente lo que es el mecanismo de plegamiento de una proteína. El plegamiento de una proteína es el proceso físico a través del cual estas biomoléculas se ordenan (y reordenan) en su estructura tridimensional característica. La predicción del plegamiento de una proteína es, en esencia, la búsqueda de la conformación funcional biológicamente activa y estable (conocida como estado nativo). Cada proteína se conforma como una cadena polipeptídica generada originalmente a partir de la información contenida en una secuencia del mRNA como cadena lineal de aminoácidos. Partiendo de esta descripción, dicho polipéptido carece de una estructura tridimensional funcional, y aunque cada aminoácido que conforma la cadena polipeptídica contiene características químicas particulares, éstas se ven afectadas por su entorno, debido a su posición en la cadena y a los vecinos espaciales. Estas interacciones pueden transformar significativamente la naturaleza química inherente de un residuo, comparada con aquella que posee cuando se encuentra aislado. Ahora bien, lo que permite al polipéptido adoptar una estructura tridimensional bien definida es el ensamble supramolecular de cada uno de los aminoácidos simples y el conjunto de interacciones con todos sus vecinos involucrados en la secuencia (Lesk, 2004 y 2009). Como ejemplo, en la figura 1 se muestra la conformación nativa de la albúmina, la principal proteína de la sangre y la más abundante en el ser humano.
El concepto esencial introducido por Levinthal es que el plegamiento de las proteínas es un problema de búsqueda al azar. Lo anterior significa que todas las conformaciones de la cadena del polipéptido (excepto la característica del estado nativo) tienen la misma probabilidad de formarse, pero el estado nativo únicamente puede hallarse por una búsqueda al azar. Levinthal era consciente de que las proteínas se pliegan de forma espontánea y en un tiempo relativamente corto, por lo que, era imposible una búsqueda aleatoria de la conformación activa de entre todas las posibles. Previo a Levinthal, Christian B. Anfinsen sugirió que la estructura de las proteínas podría predecirse a partir de la secuencia de aminoácidos (Anfinsen et al., 1961 y 1973). Su idea, conocida como la “hipótesis de Anfinsen”, es que al estimar la suma de todas las interacciones interatómicas en un espacio completo de conformaciones, es posible localizar aquella de menor energía. Anfinsen y colaboradores mostraron que las proteínas se pueden desplegar reversiblemente, implicando que las estructuras nativas de algunas proteínas se encuentren en los estados termodinámicos estables, por lo tanto, la conformación más estable es el mínimo global sobre la superficie de energía libre accesible. Levinthal, sin embargo, sugirió que la estructura estable podría ser alguna que no necesariamente se encuentra en el mínimo absoluto, lo cual es factible si la estructura de más baja energía no es cinéticamente accesible. Una analogía interesante es la de una roca rodando por una ladera, la cual se detiene en una hendidura del camino en lugar de llegar hasta la base del barranco (Hunter, 2006); en otras palabras, la roca se quedará estancada en un mínimo local. Levinthal señaló que “el plegado de proteínas es acelerado y guiado por la formación rápida de interacciones locales que determinan el futuro del plegado de la cadena; esto sugiere secuencias de aminoácidos locales que forman interacciones estables y que sirven como puntos de nucleación en el proceso de plegado” (Rooman et al., 2002). Así, Levinthal concluyó que las proteínas deben plegarse por esas “rutas” específicas.
Levinthal planteó la paradoja de plegamiento basado en dos opciones mutuamente excluyentes que se conocen como control termodinámico y control cinético del plegado de una proteína. El control termodinámico se refiere a que una proteína encuentre espontáneamente su mínimo global de energía y que el proceso de plegamiento es independiente de la ruta, esto es, la estructura nativa se determina únicamente por las condiciones finales de la proteína nativa y no por las condiciones iniciales de la proteína desplegada ni por las estructuras intermedias que se adopten en el proceso. No obstante, lo anterior le tomaría a la proteína un tiempo largo para alcanzar el estado nativo, ya que requeriría una búsqueda extensiva de una enorme cantidad de conformaciones posibles. Por otra parte, el control cinético se refiere a que el plegamiento se realiza de manera rápida, en escalas de tiempo biológicas, ya que ésta es una ruta dependiente del punto inicial, es decir, la estructura final puede diferir en función de las condiciones iniciales de la proteína desplegada desde las cuales inicia el proceso de plegamiento, por lo tanto, el estado nativo puede alcanzarse o encontrarse en un mínimo local de energía, sin necesidad de búsquedas exhaustivas.
Quizás una de las claves más sólidas para tratar de resolver esta paradoja fue propuesta por Dawkins (Dawkins, 1987) en El relojero ciego, en donde se plasma una discusión sobre la evolución por la acumulación de pequeños cambios. Dawkins tomó el comentario de Hamlet a Polonius “Methinks it is like a weasel” (creo que se parece a una comadreja) y se preguntó cómo a partir de sus componentes más simples, letras y espacios, se puede generar una frase con cierto sentido. Obviamente, el ejercicio mental debía ser un reto, por lo cual, involucró a un simio para teclear dicha frase (figura 2). La frase contiene 28 caracteres, incluyendo cinco espacios. Así, existen 27 posibilidades para localizar al azar 26 letras del alfabeto inglés y un espacio en cada una de las 28 posiciones. Un simio, entrenado para teclear al azar en una máquina de escribir, quizá requiera cerca de 2728 ≅ 1040 golpes. Dawkins planteó una segunda opción donde ahora el simio no cambiase las letras una vez que ellas se encuentren en el lugar correcto, lo que le permitiría escribir la frase empleando sólo unos miles de golpes a las teclas. La diferencia primordial es que en el primer caso se emplea una búsqueda completamente aleatoria, mientras que en la segunda, las frases intermedias del proceso, con letras parcialmente correctas, se identifican como tales y se guardan en una memoria virtual. Así, en analogía a este planteamiento, la esencia del plegamiento proteico es la retención de los intermediarios parcialmente correctos.

No obstante, el problema del plegamiento proteico es mucho más complejo que el planteado por Dawkins. En primer lugar, el criterio de exactitud (letras correctas) no se deriva de una búsqueda residuo por residuo, sino de la energía libre total de las especies transitorias, que implica tanto contactos locales en la secuencia como otros contactos cercanos en el espacio entre aminoácidos que se encuentran en posiciones lejanas en la cadena. Segundo, las proteínas no son entes del todo estables. La diferencia de energía libre entre los estados plegados y desplegados de una proteína típica de 100 residuos es del orden de 10 kcal/mol, es decir, cada residuo sólo contribuye con 0.1 kcal/mol. Dicha cantidad es menor que la energía térmica, que es de aproximadamente 0.6 kcal/mol a temperatura ambiente. Más aún, la estabilidad de los estados parcialmente plegados es menor todavía. Lo anterior significa que las diferentes conformaciones adoptadas al inicio del plegamiento podrían perderse. Así, la clave principal de este problema es que el tiempo de plegamiento real ocurre a la par del tiempo de síntesis, el cual es mucho menor que aquel necesario para explorar todas sus conformaciones, acotando así el espacio de búsqueda.
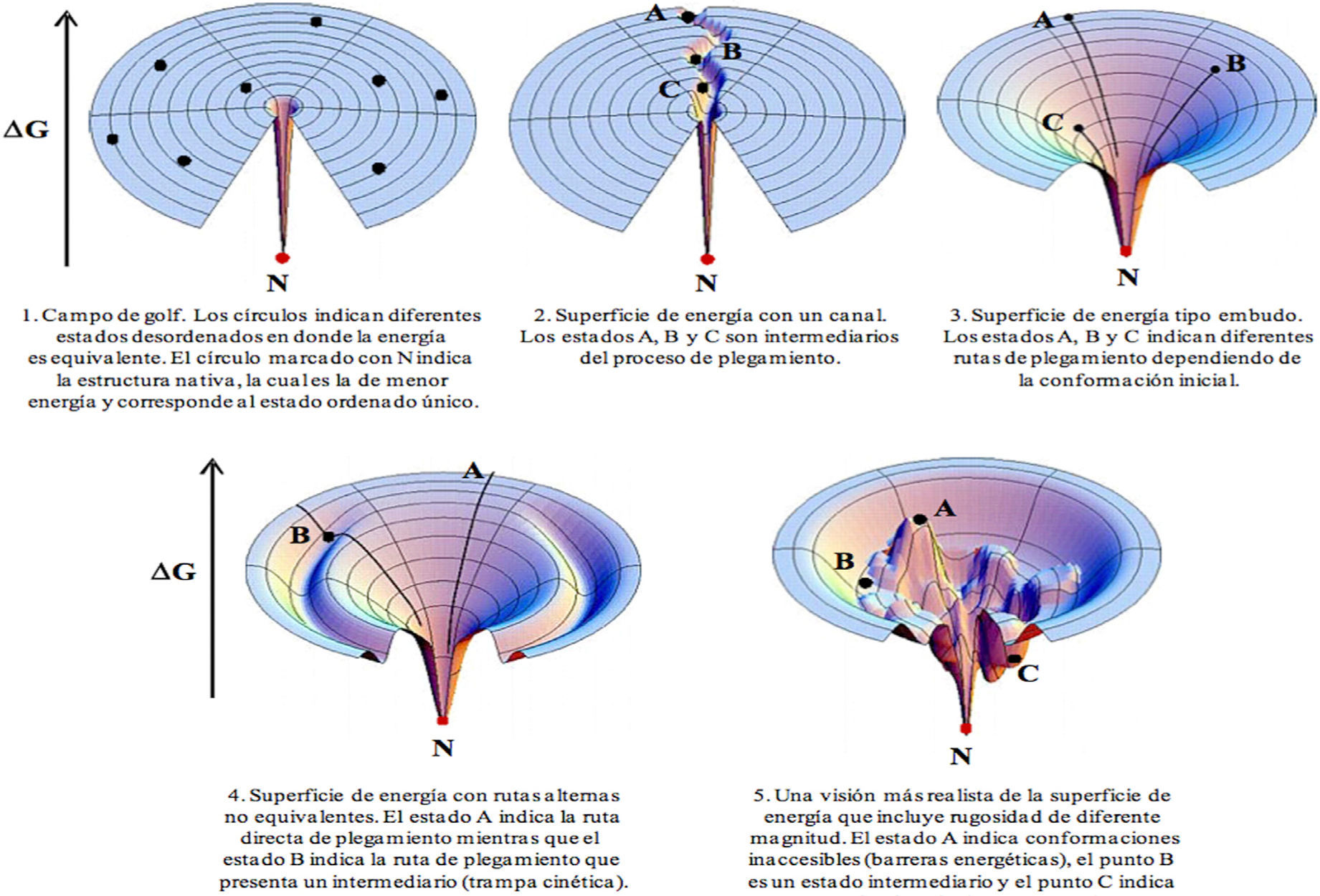
Una forma de superar la paradoja de Levinthal sería la existencia de una ruta definida de plegamiento que pasara obligatoriamente por determinados estados (intermediarios), donde cada uno de los cuales tuviera menor energía que el anterior hasta llegar al estado nativo (figura 3). Experimentalmente se han detectado algunos intermediarios del proceso de plegamiento. En particular, en varias proteínas se ha observado una estructura compacta (globular) que retiene un elevado contenido de las conformaciones locales (estructura secundaria nativa), pero que no mantiene las interacciones de rango mayor (estructura terciaria). A dicha estructura se la conoce como estado de glóbulo fundido.
.")
Diferentes tipos de superficie de energía que pueden explicar el proceso de plegamiento de una proteína. Esta figura está adecuada del manuscrito de Dill and Chan (Dill, 1997).
En la actualidad los datos experimentales sugieren que la ruta de plegamiento en la mayoría de las proteínas no es única. Por ejemplo, se ha observado que un gran número de proteínas pueden desnaturalizarse y renaturalizarse sin la presencia de algún factor o coadyudante que les permita tal reversibilidad, o en todo caso se establecen condiciones adecuadas de pH, temperatura, fuerza iónica, concentración de algún ligando, etc. Pero recientes descubrimiento han mostrado que las cosas cambian cuando el plegamiento de las proteínas ocurre in vivo, pues en ese caso, numerosas proteínas facilitan el plegamiento, como las chaperonas, las rotamasas y las disulfuro-isomerasas. Pero esto no cambia en nada en que las proteínas posean la información necesaria para buscar su conformación nativa aun estando solas. En sí, lo que hacen las proteínas que facilitan el plegamiento es catalizar, es decir, hacer más rápido y eficiente el plegamiento, mejorando la cinética y evitando la formación de estructuras no nativas. El hecho de que una proteína se pliegue por la ayuda de otras proteínas, o por factores coadyuvantes o de manera aislada, hace que la paradoja de Levinthal sea en esencia más exquisita.
Teniendo en cuenta estos datos se ha propuesto una solución alternativa a la paradoja de Levinthal. Ya que la energía de plegamiento de una proteína se asemeja a un embudo (figura 3, 3-5) y que la proteína se pliega por rutas diferentes dependiendo de la conformación en la que se encuentre en el estado desplegado, entonces cada ruta seguirá la línea de menor energía desde esta conformación hasta la estructura nativa (Dill y Chan, 1997). Una imagen ilustrativa de este proceso es la forma en que las gotas de agua alcanzan el fondo de un embudo siguiendo las líneas de máxima pendiente y sin necesidad de recorrer toda la superficie del embudo (figura 3, 3).
Hasta la fecha la paradoja aún sigue en pie pues no se conoce el proceso exacto mediante el cual se produce el plegamiento de una proteína. Se admite que este proceso se inicia con interacciones de corto alcance que forman estructuras secundarias en regiones locales del polipéptido. Estas interacciones no covalentes se establecen entre las cadenas laterales próximas. Algunos residuos tienen tendencia a formar estructuras en hélice-alfa, hoja-beta o giros reversos. Estos residuos se conocen como sitios de iniciación. El siguiente paso es la formación del estado de glóbulo fundido, el cual se produce tras un colapso hidrófobo y contiene la mayor parte de las estructuras secundarias presentes en la estructura nativa, pero posee muchas interacciones incorrectas. Las interacciones de medio y largo alcance se forman mediante reordenaciones del estado de glóbulo fundido. Las últimas interacciones en formarse, en caso de existir, son los puentes de disulfuros.
En conclusión, la paradoja de Levinthal ha adquirido cierta lógica. La búsqueda por una respuesta definitiva ha provocado la propuesta de nuevas teorías e incluso la generación de nuevos algoritmos para predecir la estructura activa de una proteína. Quizás en los próximos 10 años el avance en métodos matemáticos de localización de mínimos globales y en las técnicas experimentales de caracterización de proteínas permita resolver una de las paradojas más exquisitas y fundamentales en la Bioquímica.
S. J. Alas-Guardado agradece a conacyt por la beca otorgada durante su estancia posdoctoral en el año 2009 de acuerdo al marco de la Convocatoria “Estancias Posdoctorales y Sabáticas Vinculadas al Fortalecimiento de la Calidad del Posgrado Nacional, 2008”. Este proyecto fue financiado por conacyt M05-S01 y 105532 y daip-ugto.