


Nucleic acids and proteins comprise a network of biomacromolecules that store and transmit information sustaining the life of the cell. The study of these mechanisms is a field called molecular biology. The development of this science has always been paired with technical advances that allow breaking through methodological barriers in order to test novel hypothesis. Among available methods for molecular biologists, five stand: electrophoresis, sequencing, cloning, blotting and polymerase chain reaction. Their impact reaches genetics, medicine and biotechnology. Here, the historic relevance, technical grounds and current trends of these five essential methods are reviewed. The review intends to be useful both for students and professional scientists who seek to acquire advanced knowledge on the value of these methods to probe the molecular mechanisms sustaining life.
La biología molecular ha revelado en el último siglo varios de los mecanismos fundamentales que sustentan el funcionamiento de la célula. Hoy en día, el papel de las biomoléculas poliméricas -el ácido desoxirribonucleico ADN), el ácido ribonucléico (ARN) y las proteínas- en la salud y en las aplicaciones biotecnológicas se estudia en todos los niveles educativos. Algunos de los conocimientos más populares derivados de la biología molecular son la codificación química de información en el ADN, la secuenciación del genoma humano, la posibilidad de las terapias génicas, el diagnóstico molecular de enfermedades y la clonación de genes. Estos conocimientos no solo han sido de alto impacto científico y tecnológico, también han fascinado tanto negativa como positivamente al imaginario colectivo (Maio, 2006).
A pesar de que han pasado 84 años desde que los cimientos de la biología molecular se levantaron a partir del trabajo de Frederick Griffith (1928), todavía es difícil y puede ser polémico definir a la biología molecular. Lo anterior se debe a que es una de las ramas de la biología más dinámicas en cuanto a la cantidad y profundidad del conocimiento que ha generado y a que, frecuentemente, sus fronteras se entrelazan con ramas centenarias de la biología como la bioquímica, la microbiología y la genética.
Con base en nuestra experiencia, en este trabajo se define a la biología molecular como el estudio de los mecanismos (geométricos, mecánicos, atómicos, temporales y teóricos) mediante los cuales las moléculas participan en los procesos de transmisión de la información genética. Nuestra definición empírica está apoyada por estudios en el campo de la filosofía de la ciencia donde también se ha concluido que los tres principales conceptos que sostienen a la biología molecular son: mecanismo, gen e información (Darden y Tabery 2010). El mecanismo del flujo de la información codificada en el ADN hacia la proteína se puede analizar en la biología molecular, de forma abstracta y experimental, independientemente del flujo de materia y de energía, que se estudia de manera más adecuada por la bioquímica (Darden, 2006). Lo anterior no quiere decir que ambas áreas sean inmiscibles o que no estén conectadas, al contrario, cada vez es más frecuente que compartan métodos y conceptos para complementarse.
Los descubrimientos de la biología molecular y sus ramificaciones hacia la medicina, la biología celular y la biotecnología no se pueden entender sin el avance de las herramientas de análisis, síntesis y modificación de las moléculas de la vida. Entre los múltiples métodos de los que se vale el biólogo molecular, cinco han sido pilares en el desarrollo de la biología molecular y son parte esencial de la investigación moderna. Estos métodos son: electroforesis, secuenciación, clonación, hibridación y reacción en cadena de la polimerasa.
Dada la importancia de la biología molecular en los últimos años, el objetivo de este trabajo es presentar al estudiante que se aproxima por primera vez a esta ciencia un contexto histórico de los métodos primordiales y sus fundamentos técnicos básicos. Finalmente, para el docente y el científico profesional también se analizan sus desarrollos modernos.
2Antecedentes históricosEl evento fundacional de la biología molecular fue el reporte del “principio transformante”, un agente químico que contiene la propiedad patogénica de las bacterias de la neumonía, siendo capaz de “transformar” bacterias no patógenas en patógenas (Griffith, 1928). Ala par que se postulaban novedosas hipótesis como la del “principio transformante”, era también necesario avanzar en metodologías que permitieran un análisis científico y minucioso de las moléculas químicas en los procesos celulares. De interés especial fue el análisis de las biomoléculas de alto peso molecular o biomacromoléculas. Las primeras biomacromoléculas en estudiarse fueron las proteínas. En este contexto fue que se diseñó una técnica que permitiera separar de forma controlada a las proteínas: la electroforesis (Tiselius, 1937).
La extensión del trabajo de Griffith por Oswald Avery y sus colaboradores (Avery et al., 1944) demostró que el “principio transformante” era el ácido desoxirribonucleico (ADN). Esto colocó al ADN en el centro del entendimiento más íntimo de la célula: la naturaleza de las instrucciones que dictan su funcionamiento. De forma natural, la técnica primordial de la electroforesis fue adaptada al análisis del ADN (Thorne, 1966).
Al demostrarse la relevancia biológica del ADN y su carácter polimérico (Watson y Crick, 1953), fue necesario desarrollar métodos para conocer la secuencia de sus monómeros. Frederick Sanger fue quien desarrolló los dos métodos que extendieron a todo laboratorio de biología la posibilidad de secuenciar biomoléculas. El método de secuenciación de proteínas de Sanger (Sanger y Thompson, 1953) ha sido sustituido por el de Edman (Edman, 1950), pero el método de secuenciación de ADN de Sanger con nucleótidos terminales está ampliamente establecido como un estándar (Sanger et al., 1977). Gracias a estos métodos, se demostró la colinearidad entre el ADN y la proteína (Guest y Yanofsky, 1966). La secuenciación de biomacromoléculas se usa desde para la verificación rutinaria de moléculas recombinantes hasta para lograr el conocimiento del contenido genético total (genómica) de un organismo como el humano y otros organismos modelo. La evolución de la secuenciación de ácidos nucleicos tiene grandes alcances, por ejemplo, el análisis de bajo costo de genomas individuales con fines médicos (Ríos et al., 2010).
Otro avance importante fue el descubrimiento de las enzimas de restricción (ERs). Las ERs existen de forma natural en los microorganismos y tienen la capacidad de cortar moléculas de ADN en sitios precisos (Kelly y Smith, 1970). Con el hallazgo de las ERs de sitios específicos se fundaron las bases de la genética molecular, donde las características de un organismo ya no sólo pueden ser observadas de forma macroscópica, sino que pueden ser analizadas al nivel molecular buscando patrones de corte (huellas genéticas) de ERs en el ADN (Gusella et al., 1983). Las ERs también permitieron el desarrollo de la tecnología del ADN recombinante que consiste en clonar y expresar segmentos de ADN individuales. Así se logró el mapeo de enfermedades genéticas (Huntington’s Disease Collaborative Research Group, 1993), el descubrimiento de genes para mejorar la producción de alimentos en animales y plantas, y la producción de proteínas de alto valor en organismos de rápido crecimiento (Bolívar-Zapata, 2004).
El avance técnico individual más famoso es la invención de la Reacción en Cadena de la Polimerasa (PCR en inglés) (figura 1). La PCR es un método que simula in vitro el proceso de la replicación del ADN (Saiki et al., 1988). El método de la PCR se convirtió en una herramienta estándar mundial de la biología, ya que permite la síntesis dirigida de millones de copias de moléculas de ADN en minutos (Schmittgen y Livak, 2008). Así, el estudio del ADN ya no está restringido a las concentraciones o a sus propiedades químicas naturales. La PCR impacta muchas áreas de la ciencia y la tecnología: desde las aplicaciones forenses hasta la producción rápida de vacunas contra nuevas epidemias (Bartlett y Stirling, 2003).
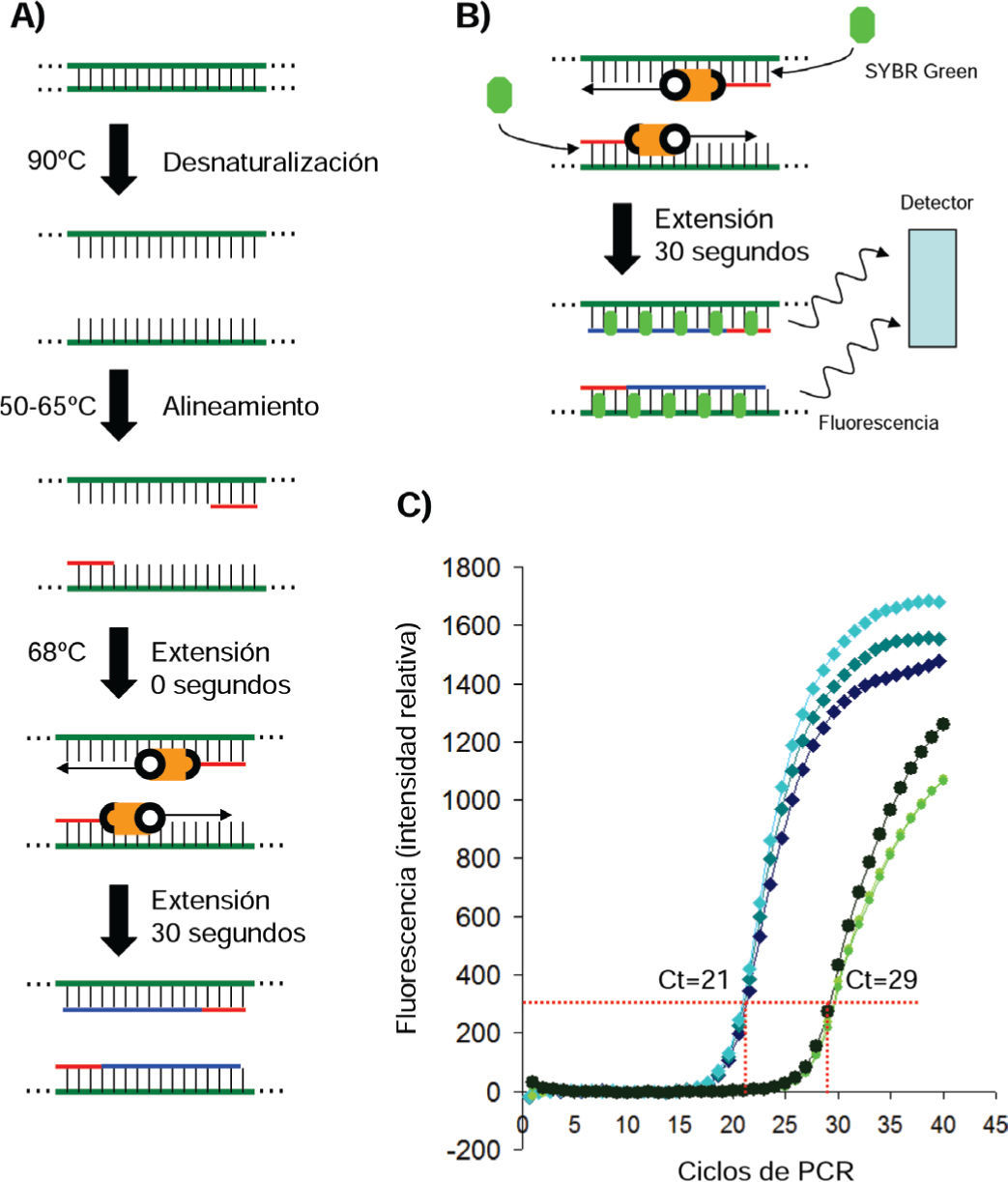
. A) Pasos térmicos de la PCR. Verde, cadena original de ADN. Rojo, oligonucleótidos. Naranja, polimerasa termoestable. Azul, nueva cadena. B) En la qPCR la cantidad de SYBR Green intercalado es directamente proporcional a la concentración de ADN. C) Experimento de qPCR. Azul, medición de actina. Verde, medición de un gen de floración vegetal (FT). Se usa el valor de fluorescencia en la etapa exponencial (ej. 300) para determinar el ciclo (Ct) en el que se detectó el mismo valor en las dos muestras. Así, Ct(actina) = 21 y Ct(FT) = 29. Si se restan los Ct resuIta 8. Dado que en cada ciclo se obtiene el doble de producto que en el anterior, se aplica 28= 256. La expresión de FT es 256 veces menor que la de actina.")
Reacción en Cadena de la Polimerasa (PCR). A) Pasos térmicos de la PCR. Verde, cadena original de ADN. Rojo, oligonucleótidos. Naranja, polimerasa termoestable. Azul, nueva cadena. B) En la qPCR la cantidad de SYBR Green intercalado es directamente proporcional a la concentración de ADN. C) Experimento de qPCR. Azul, medición de actina. Verde, medición de un gen de floración vegetal (FT). Se usa el valor de fluorescencia en la etapa exponencial (ej. 300) para determinar el ciclo (Ct) en el que se detectó el mismo valor en las dos muestras. Así, Ct(actina) = 21 y Ct(FT) = 29. Si se restan los Ct resuIta 8. Dado que en cada ciclo se obtiene el doble de producto que en el anterior, se aplica 28= 256. La expresión de FT es 256 veces menor que la de actina.
Cuatro de estos métodos tienen en común que sus inventores han sido galardonados con el premio Nobel por desarrollarlos: Arne Tiselius en 1948 (Química), Frederick Sanger en 1958 y nuevamente en 1980 (Química), Hamilton Smith en 1978 (Fisiología y Medicina) y Kary Mullís en 1993 (Química).
3ElectroforesisLa electroforesis es un método de separación basado en la movilidad de las biomoléculas en una fase líquida sometida a un campo eléctrico. Las moléculas que posean carga negativa migrarán hacia el polo positivo de un aparato electroforético y viceversa. La inclusión de una matriz sólida, además de la fase líquida, permitió agregar un nuevo punto de separación y versatilidad en la electroforesis (Smithies, 1955). De esta manera, no solo las biomoléculas pueden ser separadas por su carga, sino también por su tamaño. La inclusión de una matriz permite algo que invariablemente sorprende a los estudiantes de biología molecular: recuperar físicamente a la biomolécula que ha sido separada en un gel, con la ayuda de una navaja y un agente de visualización.
El aparato electroforético moderno tiene los siguientes componentes: un contenedor que evite la contaminación y derramamiento de las biomoléculas, dos polos que atraerán a las biomoléculas, una fuente de poder generadora de campo eléctrico regulable, una matriz que permita un segundo punto de resolución y finalmente, una solución amortiguadora de pH que mantenga la integridad de las biomoléculas (Brody y Kern, 2004).
Algunas aplicaciones electroforéticas de alta resolución requieren que el aparato pueda ser enfriado para evitar la formación de corrientes. Otros tienen sistemas de observación en tiempo real (Invitrogen, 2008a) o sistemas ópticos automatizados (Agilent, 2007). Algunos más han sido adaptados para no solamente realizar electroforesis sino también para transferir las biomoléculas que se separaron en la matriz a una superficie donde puedan ser fijadas. Este método se conoce como electrotransferencia y se emplea para aplicaciones de hibridación en membrana (ver sección 6;Bio-Rad, 2008).
3.1Electroforesis de ácidos nucleicosLa aplicación de la electroforesis en la separación de ácidos nucleicos permite hacer tareas simples, como verificar su síntesis o integridad, y complejas, como seguir los pasos en-zimáticos de modificación durante la construcción de elaboradas colecciones de ADN. Su fundamento químico reside en que los ácidos nucleicos son polímeros de carga negativa unidos por enlaces covalentes fosfodiéster. De esta manera, el ADN y el ARN se moverán en un campo electroforético hacia el polo positivo.
La matriz sólida preferida para separar los ácidos nucleicos es la agarosa pues da una capacidad de separación adecuada para la mayoría de las actividades rutinarias. Además, puede ser flexibilizada con la concentración utilizada. La electroforesis en agarosa es accesible debido a su bajo costo, a la diversidad de diseños de cámaras, y a que es fácil de preparar (Brody y Kern, 2004). El uso de acrilamida como matriz sólida, en lugar de la agarosa, aumenta la resolución a la escala de pares de bases y se aplica en el monitoreo de los pequeños ARNs, la separación de moléculas durante la secuenciación y en la separación de moléculas de pesos moleculares similares pero diferente composición de nucleótidos en gradientes desnaturalizantes (Stellwagen,2009; Barrera-Figueroa et al., 2011).
El ARN necesita un agente desnaturalizante en la matriz debido a que al ser de cadena sencilla puede hibridarse consigo mismo modificando su tamaño molecular aparente (Gerard y Miller, 1997). Frecuentemente se usa formaldehído o urea como agente desnaturalizante para romper los puentes de hidrógeno intramoleculares. Por otra parte, la visualización de los ácidos nucleicos en la electroforesis necesita de un agente. Estos son generalmente compuestos químicos de estructura plana que se intercalan entre los enlaces de hidrógeno de los ácidos nucleicos, por ejemplo, bromuro de etidio o SYBR green. Cuando estas moléculas se excitan con luz ultravioleta emiten fluorescencia que indica la posición de los ácidos nucleicos, la cual está en función de su tamaño molecular (Zipper et al., 2004).
3.2Electroforesis de proteínasLas proteínas son biomoléculas cuyos monómeros son aminoácidos polimerizados por medio de enlaces peptídicos. A diferencia de los ácidos nucleicos, que poseen una carga neta negativa, la carga de las proteínas es muy variable porque en ellas puede haber aminoácidos tanto negativos como positivos. De esta manera, las primeras separaciones electroforéticas proteicas eran toscas y necesitaban un gradiente de pH que las atrapara en su punto isoeléctrico (punto donde la proteína tendrá una carga neta de cero y no se podrá mover más en un campo eléctrico) (Tiselius, 1937). Laemmli (1970) implementó un tratamiento para poder separar a las proteínas con base en su peso molecular. Este consiste en la homogenización de la carga con dodecilsulfato de sodio (SDS), un detergente que simultáneamente desnaturaliza a las proteínas y las cubre de una carga neta negativa para que migren hacia el polo positivo durante la electroforesis. Este método se conoce como SDS-PAGE (Polyacrylamide Gel Electrophoresis). SDS-PAGE o electroforesis 1D (de una dimensión) es útil para las aplicaciones rutinarias como el seguimiento de los niveles de acumulación de proteínas, así como para su purificación, separación, hibridación con anticuerpos y análisis de peso molecular.
La electroforesis en dos dimensiones (2D) consiste en someter a las proteínas al campo eléctrico en presencia de un gradiente de pH (usualmente 2-11) que enfocará a las proteínas en su punto isoeléctrico. El segundo paso consiste en tratar al gel con SDS, colocarlo en un ángulo de 90° en relación a su posición original y realizar una electroforesis 1D para separar por peso molecular (Garfin, 2003). El método 2D puede separar a prácticamente todas las proteínas presentes en una condición biológica, mismas que pueden ser secuenciadas e identificadas mediante espectrometría de masas (MS). Cuando la electroforesis 2D se combinó con el método analítico de espectrometría de masas (MS), se fundó una nueva rama del estudio masivo de las proteínas que se conoce como proteómica (Yates, 2011). Ambos tipos de electroforesis de proteínas son visualizados con tinciones como la de azul de Coomassie o con plata (Sambrook et al., 2001).
3.3Avances en la electroforesisEl aparato electroforético más miniaturizado es el Bioanalyzer 2100 (Agilent, 2007). Esta máquina realiza la electroforesis en matrices de tan solo 1 × 2 mm (a diferencia de las matrices tradicionales que pueden medir varios centímetros) con gran resolución gracias a un sistema óptico-digital de lectura. El aparato electroforético más automatizado es QIAxcel (Qiagen, 2008). Está basado en microelectroforesis en capilares y permite la visualización de los datos en tiempo real. Se puede realizar el procesamiento de miles de muestras de forma automática. Los costos elevados de estos aparatos son prohibitivos para laboratorios individuales, pero se les encuentra en instalaciones compartidas.
El reto actual de la electroforesis de proteínas está en su acoplamiento automático con métodos río abajo, especialmente con MS que permita identificar a las proteínas separadas e intactas (Tran et al., 2011). Adicionalmente, se está buscando la estandarización para construir bases de datos de imágenes electroforéticas 2D de las más diversas condiciones biológicas y experimentales (Bland et al., 2010), así como las herramientas de cómputo (bioinformática) para extraer la mayor información biológica posible de estos experimentos (Boyle et al., 2012).
4SecuenciaciónADN, ARN y Proteínas forman el orden del flujo de la información genética en todas las células y los tres son polímeros lineares. El objetivo de la secuenciación es conocer exactamente el orden de sus monómeros. Del orden de los nucleótidos en el ADN y ARN se puede inferir su evolución y su función (al menos en teoría), lo mismo ocurre con los aminoácidos de las proteínas.
4.1Secuenciación de ácidos nucleicosDe todos los métodos históricos de secuenciación de ácidos nucleicos diseñados por Sanger, el método por terminadores dideoxi es el más exitoso (Sanger, 1988). Este método usa nucleótidos modificados con la ausencia del extremo 3’OH, grupo esencial para la polimerización por la ADN Polimerasa (figura 2A). De esta forma, cuando se incorpora un dideoxinucleótido, queda un trozo trunco de ADN que puede ser separado electroforéticamente (Sanger et al., 1977). Si cada uno de los cuatro monómeros de ADN (los nucleótidos adenina, guanina, citosina y timina) se marca con una molécula fluorescente diferente, se puede determinar rápidamente el orden de los nucleótidos. La secuenciación con el método dideoxi se provee usualmente en centrales de secuenciación a muy bajo costo. La secuenciación dideoxi acoplada a métodos de reconstrucción computacional (Shotgun) ha sido fundamental en la secuenciación de los genomas modelo de humano, animales y plantas (Galperin y Koonin, 2010).
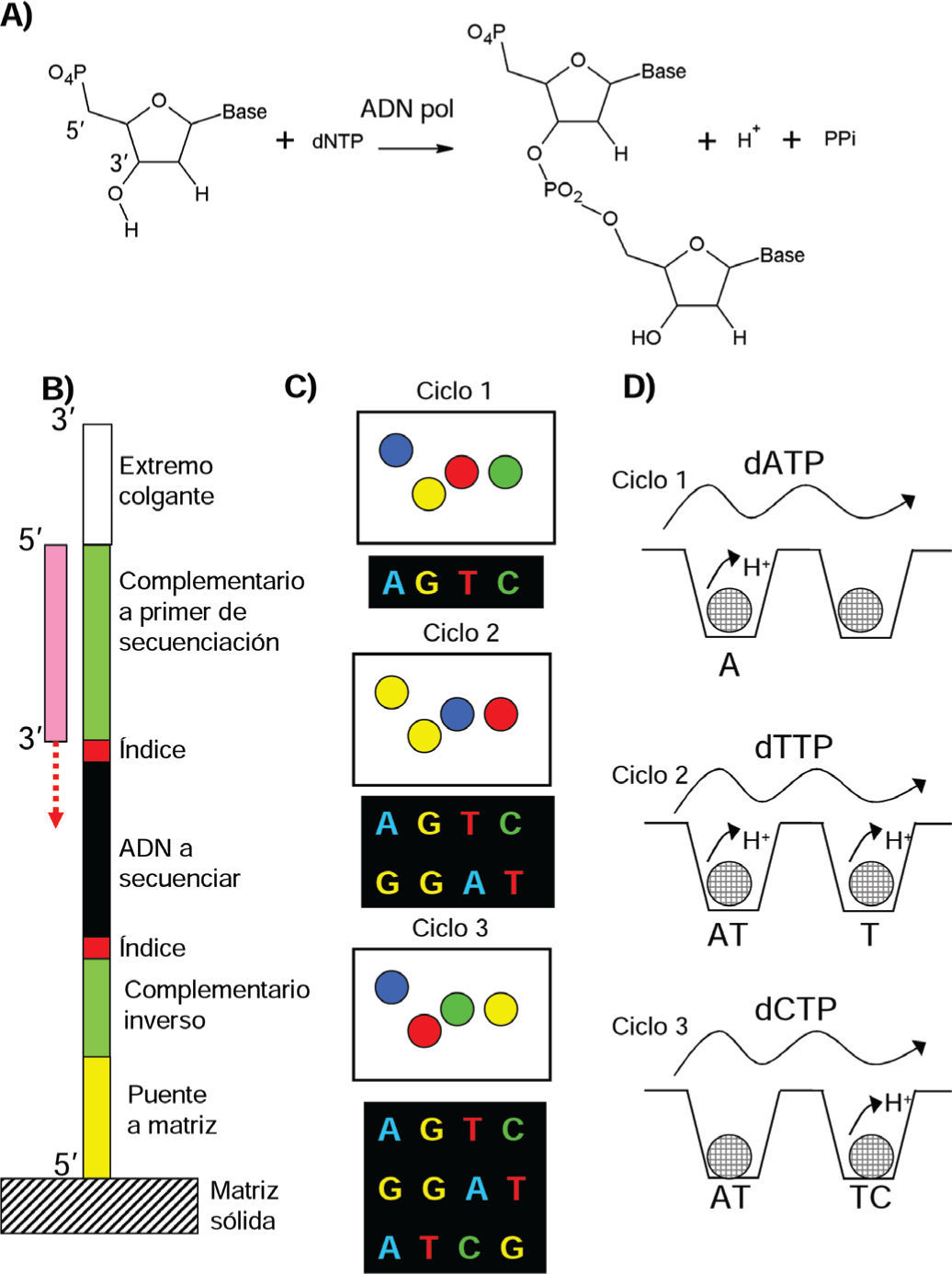
. A) Reacción catalizada por la ADN Polimerasa. La adición de un nuevo nucleótido (dNTP) al extremo 3’ OH de la molécula resulta en la liberación de pirofosfato y un protón. B) Las plataformas de secuenciación procesan quimeras de ADN. C) Secuenciación por síntesis y observación microscópica de fluorescencia (Illumina). Durante cada ciclo se añade un nuevo nucleótido fluorescente que detiene la síntesis, se toma una micrografía de la matriz de secuenciación, se modifica el último nucleótido para que pueda aceptar uno nuevo y se pasa al siguiente ciclo. El análisis digital de las micrografías determinara la secuencia. D) Secuenciación por detección de incrementos de pH (Ion Torrent). En cada ciclo se baña la matriz con un solo nucleótido, los pozos donde se integre a la cadena de Adn registrarán un incremento transitorio de pH por la liberación del protón.")
Secuenciación masiva post-Sanger de última generación (por síntesis). A) Reacción catalizada por la ADN Polimerasa. La adición de un nuevo nucleótido (dNTP) al extremo 3’ OH de la molécula resulta en la liberación de pirofosfato y un protón. B) Las plataformas de secuenciación procesan quimeras de ADN. C) Secuenciación por síntesis y observación microscópica de fluorescencia (Illumina). Durante cada ciclo se añade un nuevo nucleótido fluorescente que detiene la síntesis, se toma una micrografía de la matriz de secuenciación, se modifica el último nucleótido para que pueda aceptar uno nuevo y se pasa al siguiente ciclo. El análisis digital de las micrografías determinara la secuencia. D) Secuenciación por detección de incrementos de pH (Ion Torrent). En cada ciclo se baña la matriz con un solo nucleótido, los pozos donde se integre a la cadena de Adn registrarán un incremento transitorio de pH por la liberación del protón.
La secuenciación de proteínas mediante el método de Ed-man consiste en marcar el extremo N-terminal de las moléculas con el reactivo químico fenilisotiocianato (PITC) (Edman, 1950). La identidad del PITC-aminoácido escindido de la proteína puede ser determinada mediante la observación de su presencia directamente en cromatografía de capa fina o de su ausencia por exclusión en un analizador de aminoácidos (Carlton y Yanofsky, 1965). Pronto el método de Edman fue automatizado y es actualmente el estándar de secuenciación de proteínas. En años recientes, este método químico ha coexistido con el método analítico MALDI (matrix-associated laser desorption/ionizaton) acoplado a MS, que identifica los péptidos de acuerdo a su huella de fragmentación iónica (Lewis et al., 2000).
4.3Avances de la secuenciación de ácidos nucleicosRecientemente, la secuenciación de ácidos nucleicos se ha colocado de nueva cuenta en la frontera de la investigación. Los nuevos métodos se han alejado de la metodología de Sanger y se han movido hacia la secuenciación por síntesis catalizada por la ADN polimerasa (figura 2A). De esta manera, si a la molécula de ADN a secuenciar se le agregan, mediante pasos sencillos de ligación y PCR, nucleótidos de secuencia conocida en sus extremos para que se adsorba por hibridación a una superficie (figura 2B), las bases incorporadas al ADN se pueden detectar de manera directa, durante su síntesis, mediante marcado fluorescente diferencial de los cuatro posibles nucleótidos del ADN (figura 2C) (Bentley et al., 2008).
También se puede detectar la incorporación de las bases de manera indirecta midiendo los cambios de pH ocasionados por la producción de protones durante la polimerización (figura 2D) o midiendo la concentración del pirofosfato también producido durante la polimerización (método 454) (Rothberg et al., 2011; Margulies et al., 2005). Así, llevando a cabo estas reacciones sobre plataformas nanométricas, se pueden secuenciar simultáneamente decenas de millones de fragmentos cortos de 37-200 bases, dependiendo del método. En estos métodos se ha sacrificado la longitud del fragmento secuenciado por el número de fragmentos que se secuencian.
La secuenciación por síntesis permite la re-secuencia-ción de genomas haciendo realidad la medicina genómica personalizada donde se pueden buscar marcadores o descubrir alelos responsables de enfermedades genéticas en individuos (Ng et al., 2010). Todo esto ha sido posible gracias al diseño de ingeniosas plataformas que emplean semiconductores y nanopotenciometros, abaratando así la impresionante capacidad de secuenciación moderna (figura 2D) (Rothberg et al., 2011).
Debido a la gran capacidad de obtención de información que permiten estos métodos (en el orden de terabytes) un reto moderno muy importante es optimizar los métodos bioinformáticos necesarios para analizar estos datos y obtener de ellos la mayor cantidad de información biológica posible de manera intuitiva (Boyle et al., 2012).
5ClonaciónLa clonación de ADN fue posible gracias a la acumulación de conocimiento básico sobre las enzimas de restricción (ER). Las ER fueron descubiertas en el estudio de los mecanismos bacterianos de defensa ante las infecciones virales (Kelly y Smith, 1970). Actualmente, en los catálogos comerciales hay más de 200 ERs que se producen de forma recombinante (New England Biolabs, 2011a).
Las ERs cortan de forma precisa secciones de ADN y hacen compatibles los extremos de los productos escindidos con otras moléculas de ADN, como adaptadores, plásmidos, promotores y regiones codificantes, a los que se unen en una reacción in vitro catalizada por la enzima ligasa. Estas nuevas construcciones de ADN formadas por moléculas de orígenes genéticos distintos, se denominan quimeras. Las quimeras a su vez pueden ser escindidas para ligarse a otro fragmento de ADN, en un proceso llamado subclonación. Mediante programas de cómputo se puede simular la mejor estrategia para obtener quimeras en el laboratorio (Invitrogen, 2008b). Las quimeras son útiles para comprobar hipótesis sobre la función génica, así como para la producción en masa de proteínas recombinantes (Goeddel et al., 1979, Chan et al., 2010).
Las ERs sirvieron durante la segunda mitad del siglo xx para caracterizar ADN de tamaño muy grande (ej. 500,000 bases) imposible de manejar en su tamaño original. En combinación con las hibridaciones Southern Blot (ver sección 6), las ERs fueron esenciales para descubrir los primeros genes responsables de enfermedades genéticas humanas (Gusella et al., 1983) y para seleccionar características deseables en vegetales y animales (Bolívar-Zapata, 2004). Actualmente, debido a lo laborioso que es obtener huellas genéticas con ERs, se favorece la obtención de huellas por PCR o por secuenciación masiva del genoma completo con métodos post-Sanger (ver secciones 4 y 7; Ríos et al., 2010).
5.1Avances en la clonaciónLa investigación sobre ERs tiene una línea muy activa dirigida hacia el descubrimiento de ERs con sitios de corte novedosos, así como de ingeniería de proteínas para mejorar la velocidad de digestión o la eliminación de actividades secundarias no deseadas (Chan et al., 2010).
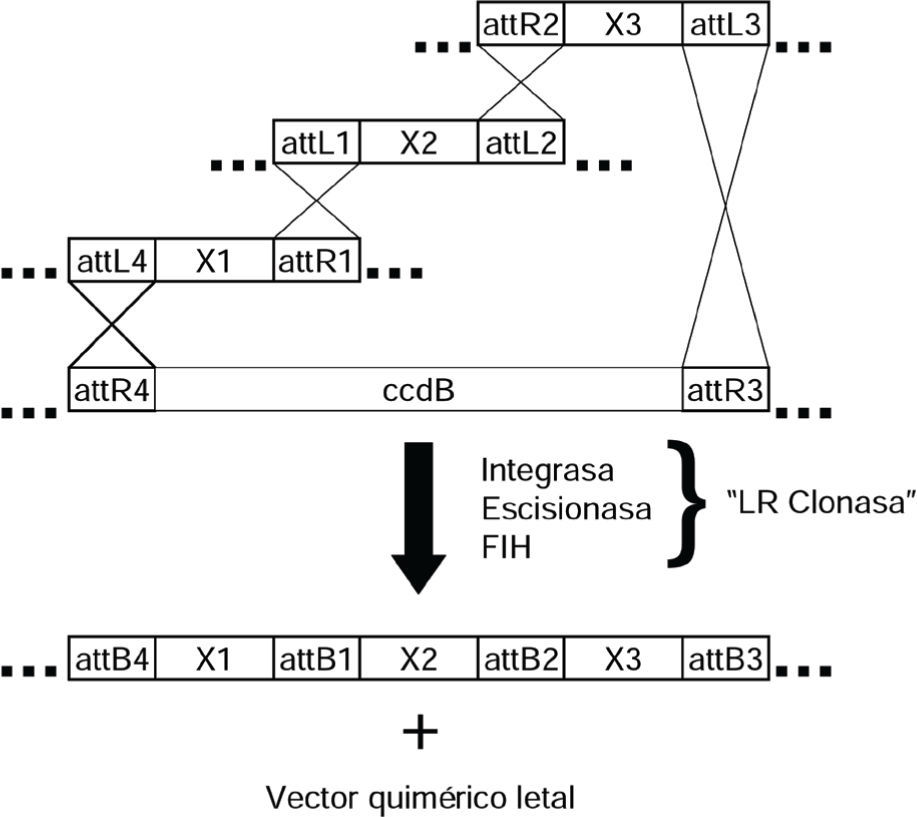
Recientemente, una reacción enzimática de clonación directional llamada Gateway permite la clonación y subclonación sin utilizar ERs (Magnani et al., 2006). Gateway explota las características de recombinación del bacteriófago lambda, incluyendo las integrasas y los sitios attX (Van Duyne, 2005). Mediante el uso de cuatros sitios diferentes (attB, attP, attL y attR), tres o más moléculas de ADN pueden ser clonadas en vectores receptores (figura 3). Nuevas tecnologías post-Gateway se están proponiendo. Una de las más prometedoras es la escisión de uracilo que une más de cinco secciones de ADN de orígenes independientes en una sola quimera sin dejar “cicatrices”, es decir, sin dejar bases extras de ADN no deseadas en el sitio de unión (Norholm, 2010).
. X denota cualquier fragmento de ADN, de esta forma, una selección típica puede ser X1 = promotor, X2 = secuencia codificante y X3 = terminador. Si el vector que contiene a ccdB incluyera el promotor o el terminador, X3 puede ser un marcador inmunológico o una proteína fluorescente de fusión para detectar al producto de X2. El producto secundario de la reacciones es un vector quimérico letal gracias al gen ccdB que codifica para una toxina que inhibe la replicación de ADN, por lo tanto, no se recupera en las operaciones río abajo.")
Tecnología de ADN recombinante libre de enzimas de restricción. Se ilustra la tecnología Gateway para la construcción de quimeras de ADN. En la secuencia de ADN de los sitios att reside su alta especificidad de recombinación. Con la catálisis de la mezcla de enzimas integrasa, escisionasa y factor de integración al huésped se logra la recombinación de tres diferentes plásmidos o productos de PCR (aunque con sitios adicionales, hasta cuatro fragmentos se oueden recombinar). X denota cualquier fragmento de ADN, de esta forma, una selección típica puede ser X1 = promotor, X2 = secuencia codificante y X3 = terminador. Si el vector que contiene a ccdB incluyera el promotor o el terminador, X3 puede ser un marcador inmunológico o una proteína fluorescente de fusión para detectar al producto de X2. El producto secundario de la reacciones es un vector quimérico letal gracias al gen ccdB que codifica para una toxina que inhibe la replicación de ADN, por lo tanto, no se recupera en las operaciones río abajo.
Cuando los ácidos nucleicos o las proteínas en solución interactúan con superficies sólidas pueden llegar a adsorberse en ellas. Las superficies más utilizadas son las membranas de nitrocelulosayde nylon. Esta propiedad de interacción no ha sido clarificada del todo pero se plantea la hipótesis de que reside en interacciones no covalentes como interacciones hidrofóbicas o de cargas parciales entre las biomoléculas y la superficie (Dubitsky y Perrault, 2012). En el caso de los ácidos nucleicos, estas interacciones se pueden hacer covalentes con luz ultravioleta. En los catálogos modernos se puede encontrar una gran variedad de membranas en cuanto a capacidad de adsorción, resistencia, tamaño y grupos químicos activos, que se pueden adecuar a diferentes aplicaciones (GE, 2011).
Esta propiedad de interacción con fases sólidas, en conjunto con la capacidad de los ácidos nucleicos de formar puentes de hidrógeno con otras moléculas de ADN complementarias a su secuencia (hibridación), se han utilizado para detectar y cuantificar su abundancia en extractos celulares de las más variadas condiciones. La técnica original se conoce, por su nombre en inglés, como Southern Blot (Southern, 1975). Una traducción aproximada sería “manchas de Southern”. Los Southern Blot consisten en adsorber una muestra de ADN (digerida por enzimas de restricción y separada previamente de manera electroforética) en una membrana, en presencia de un agente que rompa los puentes de hidrógeno intermoleculares (ej. NaOH), esto es, que desnaturalice la estructura del ADN en la membrana. Las moléculas contenidas en la muestra adsorbida en la membrana, de las cuales se busca obtener información específica, se llaman “blanco”. Posteriormente, otra molécula de ADN en solución, también desnaturalizada (por calor) o de cadena simple, se hace interactuar con el ADN en la membrana a una temperatura que permita un reconocimiento de bases lo más específico posible. Esta molécula que servirá para extraer información sobre el blanco, se denomina “sonda”. Si la sonda ha sido marcada con radioactividad o grupos antigénicos (capaces de interactuar con anticuerpos), producirá manchas oscuras en la zona de hibridación si se le expone a una placa fotográfica (Southern, 1975). El patrón de manchas producidas indicará la presencia de su blanco, y dará información sobre su tamaño, frecuencia y abundancia en la muestra.
Si en este arreglo en lugar de colocar ADN en la membrana se coloca ARN desnaturalizado con formaldehido, se le denomina Northern Blot, esto debido a un juego de palabras con el Southern Blot. Si en la membrana se colocan proteínas desnaturalizadas con SDS y en la solución anticuerpos (proteínas del sistema inmune capaces de interactuar con otras proteínas), el método se conoce como Western Blot (continuando con el juego de los puntos cardinales) (Towbin et al., 1979).
El diseño de experimentos con estos tres tipos básicos de “blots” involucra el uso de condiciones contrastantes, es decir, la comparación de las propiedades de las manchas que se obtengan (tamaño molecular, intensidad, número) de una muestra control o estándar contra las muestras donde se espera un comportamiento diferente, por ejemplo, las señales detectadas en ARN extraído de células sanas contra las detectadas en ARN extraído de células de un tumor cancerígeno. La combinación de la fuente de los extractos de biomoléculas (téjidos, células, mutantes, individuos), de sondas (genómicas, transcriptómicas, anticuerpos monoclonales o policlonales), de ERs o de temperatura de hibridación, ha permitido los más complejos análisis como la detección de los defectos moleculares donde reside la causa de una enfermedad humana (Gusella et al., 1983) o la manera como las plantas miden la longitud del día mediante la acumulación de biomoléculas (Corbesier et al., 2007), por ejemplo.
En los años 80 surgió la idea de adsorber en la membrana al ADN de las sondas y no al ADN de las muestras o blancos. Esta sencilla modificación revolucionó los métodos de descubrimiento basados en las interacciones moleculares y recibió el nombre de macroarreglo o Dot Blot (Augenlicht y Kobrin, 1982). Los macroarreglos son útiles para analizar al mismo tiempo el comportamiento de decenas o cientos de moléculas de ADN O ARN (en forma de ADNc, ver Sección 7) de secuencia conocida. La cantidad de sonda que se coloca (imprime) en un macroarreglo está en el orden de microgramos en microlitros, sin embargo, con el advenimiento de los métodos de microfluídos fue posible manejar cantidades en el orden de nano- pico y fentolitros (Kirby 2009). Al minaturizar el volumen de muestra usada, la cantidad de puntos individuales que se podían imprimir (ahora con la ayuda de robots) y por ende analizar en el mismo espacio, aumentó logarítmicamente hasta el orden de miles. Estos macroarreglos miniaturizados se han denominado microarreglos y son el fundamento técnico de la transcriptómica, es decir, el conocimiento global de la expresión de mensajeros de ARN en un organismo (Schena et al., 1995).
El extenso uso de macro- y microarreglos por la comunidad de biólogos moleculares modernos ha permitido la construcción de bases de datos internacionales gratuitas donde se depositan los análisis de este tipo de experimentos, mismos que pueden ser consultados a través de portales de internet en interfases intuitivas. Por ejemplo, la base Gene Expression Omnibus aloja actualmente casi 30,000 experimentos individuales de expresión de ARN y con más de 700,000 condiciones biológicas exploradas (Barrett et al., 2011). Desde 2010 hasta el primer cuarto de 2012 la cantidad de contribuciones a GEO han crecido en un 33% (GEO, 2012).
6.1Avances en la detección de biomoléculas en superficiesLa capacidad de impresión de ácidos nucléicos en forma de microarreglo ha aumentado exponencialmente al comenzar a utilizarse vidrio como la superficie preferida y el método de síntesis de ADN conocido como fotoquimolitografía como método de preparación. En la quimiolitografía se utilizan máscaras metálicas de tamaños micrométricos para dirigir la polimerización química del ADN previamente diseñado en computadora (Miller y Tang, 2009). Con el advenimiento de las secuenciación de organismos completos, se hizo posible diseñar sondas de ADN que cubren prácticamente todo el genoma de organismos tan complejos como el ser humano (Brady y Vermeesch, 2012). Así, hoy en día los biólogos moleculares tienen a su alcance plataformas de microarreglos estandarizadas para analizar ADN y ARN en prácticamente todos los organismos modelo del árbol de la vida. Los microarreglos y la secuenciación por síntesis han llegado al mismo punto en el que la información potencial que se extrae de los experimentos que las emplean es tanta que es un reto bioinformático analizarla (Boyle et al., 2012).
7Reacción en cadena de la polimerasa (PCR)La PCR es un método que permite la amplificación o copiado masivo in vitro de un fragmento específico de ADN. La longitud del fragmento copiado está indicada por dos secuencias pequeñas de ADN (llamadas oligonucleótidos) que son adicionadas a la reacción y que flanquean al fragmento que se desea amplificar. Estas secuencias pequeñas indican además el sitio desde el cual debe iniciarse la reacción de amplificación. La reacción es llevada a cabo por la ADN polimerasa.
Dos avances técnicos hicieron popular a la PCR. Uno fue la invención de los termocicladores (Hsieh y Chen, 2009). Los termocicladores realizan automáticamente los pasos de incremento y decremento de la temperatura indispensables en la PCR (figura 1A). El otro descubrimiento fue el de las ADN polimerasas termoestables (Chien et al., 1976). Estas polimerasas toleran las temperaturas a las que se lleva a cabo una PCR. Hoy en día las variantes de polimerasas termoestables que existen en el mercado se pueden contar por decenas. Se pueden seleccionar por velocidad de síntesis, fidelidad del copiado, capacidad de sintetizar moléculas largas, tolerancia a inhibidores de reacción y por ornamentaciones adicionales que hacen a los productos (ej. adenilación terminal) (New England Biolabs, 2011b).
Cada ciclo de PCR tiene tres pasos térmicos (figura 1A). Desnaturalización: se calienta la reacción (90-94°C) para romper los puentes de hidrógeno y separar la doble cadena de ADN. Alineamiento: se diminuye la temperatura (50-65°C) para alinear los oligonucleótidos con su molécula blanco. Extensión: se eleva la temperatura (68-72°C) para que la polimerasa termoestable busque su sustrato de doble cadena (ADN molde + oligonucleótido) y sintetice la nueva hebra de ADN utilizando a los nucleótidos libres en solución (figura 2A) (Bartlett y Stirling, 2003).
Este sencillo arreglo puede ser manipulado de las maneras más ingeniosas. Los nucleótidos pueden ser modificados con grupos antigénicos o radioactivos para detectarlos por hibridación. Los oligonucleótidos pueden ser modificados con sitios de corte de ERs para poder unir moléculas antes imposibles de ensamblar. Se pueden agregar secciones prediseñadas de ADN (adaptadores) para posteriormente amplificar o secuenciar regiones desconocidas. Se puede modificar la PCR para que la polimerasa se equivoque frecuentemente en la adición de nucleótidos para generar miles de mutaciones diferentes y simular el proceso natural de la evolución (Bartlett y Stirling, 2003).
Una adición relevante a la PCR convencional es su acoplamiento con otra reacción enzimática llamada retrotranscripción (RT). En la RT se emplea a una enzima denominada retrotranscriptasa que posee la capacidad catalítica de sintetizar ADN a partir de ARN produciendo ADN complementario o CADN (Baltimore, 1970; Temin y Mizutani, 1970). Cuando en la PCR el ADN molde es CADN, la PCR se transforma en un método de amplificación indirecta del ARN (Gibson et al., 1996).
La adición de SYBR Green como un componente de la reacción y la inclusión de un lector de fluorescencia en los termocicladores revolucionó la PCR (Bustin et al., 2009). De esta manera, cuando en la extensión se sintetizan moléculas nuevas de ADN, SYBR Green se intercala entre ellas emitiendo fluorescencia. La fluorescencia es directamente proporcional a la abundancia de moléculas sintetizadas, que a su vez es una medida directa del número de moléculas que se encontraban en la muestra al inicio de la PCR (figura 1B). Mediante el uso de estándares de concentración conocidos o el uso de métodos de comparación relativa, la PCR se vuelve un método efectivamente cuantitativo (figura 1C) (Schmittgen y Livak, 2008). Esta modificación se llama PCR cuantitativo (qPCR).
Antes del qPCR, los métodos de cuantificación de abundancia de ácidos nucleicos se basaban en la hibridación en fase sólida (Northern y Southern Blot), o en sus variantes de análisis masivo como los microarreglos. La qPCR ha sustituido a todos ellos -excepto a los microarreglos- como el método primario para cuantificar la abundancia absoluta o relativa de ADN (Russell, 2011). La qPCR requiere de una sencilla pero cuidadosa estandarización detallada en un protocolo internacionalmente aceptado (Bustin et al., 2009).
7.1Avances modernos en la PCRLa investigación sobre la PCR tiene como objetivo central ampliar las aplicaciones del método a todos los aspectos biológicos de la investigación. Desde el diagnóstico molecular de enfermedades hasta métodos de qPCR que sean capaces de detectar pequeños ARNs de 22 pb (Schmittgen and Livak, 2008). La automatización de la PCR es también una necesidad real ya que la PCR ha sustituido al mapeo genético por ERs (Sigma, 2011). De esta forma, la caracterización de bancos de mutantes requiere la preparación de miles de reacciones y de su análisis en sistemas electroforéticos de última generación.
8Fuentes de consulta de protocolosLa descripción de los protocolos paso a paso va más allá del alcance de esta revisión. Estos se pueden consultar en blogs gratutitos (protocol-online.org; e-biotek.com), en la American Society for Cell Biology (cellbase.ascb.org), y en Cold Spring Harbor Laboratories (cshprotocols.cshlp.org). También en revistas como Nature Methods, Biotechniques y Methods in Enzymology. No hay que olvidar el libro clásico Molecular Cloning (Sambrook et al., 2001).
9ConclusionesLa electroforesis, la secuenciación, la clonación, la hibridación y la PCR raramente se usan en un contexto individual. Actualmente se aplican en una cadena de análisis combinados entre ellas y con otras metodologías como extracciones, modificaciones enzimáticas y químicas, y purificación. Todo con el fin de probar hipótesis sobre el papel de las biomoléculas en el flujo de la información genética.
El costo del análisis molecular ha disminuido marcadamente, permitiendo aumentar el número de muestras experimentales. Lo anterior ha sido posible gracias a los métodos de secuenciación masiva, la robotización de los procedimientos manuales, el desarrollo de los microfluídos, la miniaturización de las plataformas de análisis y la producción estandarizada de enzimas y matrices. Estas tendencias modernas de la investigación técnica en la biología molecular no son exclusivas de los métodos aquí revisados; es algo extensivo a todos los métodos de la biología molecular. A pesar de que han pasado décadas desde que se desarrollaron estos métodos, su omnipresencia en los reportes científicos de la biología moderna se debe a que son métodos versátiles, económicos, automatizables y reproducibles.
A nuestros colegas de laboratorio de los últimos 10 años por las interesantes discusiones sobre métodos moleculares, así como a los estudiantes de biotecnología del Instituto Politécnico Nacional y de la Universidad del Papaloapan por su infinita curiosidad. También a S. del Moral, J. Abad y E.Villalobos por revisar el manuscrito preeliminar. La investigación de los autores es apoyada por los proyectos SEP-CO-NACyT (152643, 169619), SEP-PROMEP (103.5/11/6720) y Repatriación (146633).