



Molecular Visualization of the consequences of site-directed mutations can help teaching the structure-activity relationships of proteins to undergraduate and graduate students. However, experimental structural data are usually limited to a few mutations, mostly at active sites, and real-time computations are too time-consuming for standard classroom sessions. Since precalculation of all possible mutations for a given protein is impractical, a database of models with either a subtle (alanine), or aggressive site-mutations of the yeast pyrophosphatase homodimer was created. The models are displayed with the VMD package at IXTLI (virtual reality facility at UNAM). We programmed a user interface to create the illusion of real-time mutations at a mouse click. In a session, students worked with the program, under a semi-guided assignment, finding the interface friendly and learning to exploit the software quickly. Their performances varied substantially, but most were highly motivated, and seemed to have benefited from the session.
La visualización molecular de las consecuencias de mutaciones dirigidas puede auxiliar a estudiantes de licenciatura y de grado en el aprendizaje de las relaciones estructura-actividad de proteínas. Sin embargo, los datos estructurales experimentales se limitan a unas pocas mutaciones, la mayoría sobre sitios activos y los cálculos de cómputo a tiempo real son muy lentos para ser utilizados en una sesión de clase estándar. Dado que el precálculo de todas las posibles mutaciones para una proteína dada no es factible, se optó por crear una base de datos de modelos con una mutación de sitio suave (alanina) o una agresiva de la pirofosfatasa homodimérica. Los modelos se despliegan con el paquete VMD en el laboratorio IXTLI (la instalación de realidad virtual de la UNAM). Programamos una interfase de usuario para crear la ilusión de mutaciones en tiempo real con un click del ratón. En una sesión, en la que los estudiantes trabajaron semi-guiados con el programa, encontraron amigable la interfase y explotaron las capacidades del software rápidamente. Su desempeño varió sustancialmente, pero la mayoría se mostró altamente motivada y beneficiada por la sesión.
At the core of modern biochemistry, there are two central aspects: the central dogma of Molecular Biology and the principles that govern the structure-activity relationship of proteins. Both subjects involve a significant number of hard-to-learn concepts, but they differ in the kind of skills they demand from the students. The first one involves a clear understanding of cell organization, its interaction with the surrounding medium, and how living systems handle and control genetic information. On the other side, understanding the structure and functions of proteins requires good knowledge of physical molecular forces, chemical bonding, and the ability to visualize three-dimensional shapes, as well as the spatial relationships between them. Due to their complexity and the level of abstraction demanded, the latter skills are harder to acquire by the average student. Interactive molecular visualization techniques, along with the use of computational molecular models, aid the understanding and comparison of the structural basis of molecular function, in a dynamic way. Learning activities as the one presented here have been considered as teaching aids in the development of new observation and thinking skills by the students (Root-Bernstein, 2002).
Together with the publicly available 3D structures form the Protein Data Bank (PDB) and the visualization tools built therein (Berman et al., 2000; www.pdb.org), a number of friendly computer programs are available to display and manipulate macromolecular 3D structures, with relative ease, including the well known Rasmol (Sayle and Milner-White, 1995), its open-source replacement Jmol (Herráez, 2006; www.jmol.org), or the full featured PyMOL (DeLano Scientific LLC, San Carlos, CA, USA) and VMD (Humphrey et al., 1996), amongst others. These are a remarkable set of tools for teaching protein structure and function, but there are still a number of limitations, specially when it comes to endowing the students with freedom to mutate an amino acid at some protein location, and analyze its consequences. While a number of force fields have proven robust enough to provide reasonable predictions and render realistic molecular dynamics simulations, the amount of time and computing resources to complete the calculations make them impractical to use during a class session, specially if a large number of possible mutants is desired. Besides, the steep learningcurve associated to the use of simulation software packages is in most cases beyond the scope of a undergraduate biochemistry course.
To tackle these limitations, while maintaining a good degree of flexibility, we decided to select one specific protein, within the frame of a specific biochemical problem, thus constraining the number of sites accepting mutations and the possible substitutions at each position. We intended to make these restrictions appear as “intuitive” to the students, so we decided to focus on the subunit interface of a middle-sized soluble homodimer, putting aside the active-site problem, as a realistic treatment of catalysis implies very demanding quantum chemical calculations.
We choose the soluble inorganic pyrophosphatases because they are a protein family with a highly diverse set of quaternary structures along evolution (Sivula et al., 1999; Gómez-García et al., 2007). Although this paper only deals with the case of the dimeric yeast pyrophosphatase, it can be extended to more complex cases. We show here how to create the illusion of real-time mutations at a “mouse-click” using Tcl/Tk scripting (Ousterhout, 1994; Welch et al., 2003) within VMD (Humphrey et al., 1996) a freely available molecular visualization package. This software was deployed under the virtual reality environment at the IXTLI observatory of UNAM (Dirección General de Servicios de Cómputo Académico, Universidad Nacional Autónoma de México, Mexico City, Mexico; www.ixtli.unam.mx) to render a fully immersive, interactive experience. In addition, we summarize the written commentaries made by a group of students, at the end of a semiguided exercise, using the resulting tools.
The simulationIn silico generation of a realistic mutants-database The 3D structure of the yeast soluble inorganic pyrophosphatase 1E9G (Heikinheimo, et al., 2001) was obtained form the PDB. This is a middle-sized dimeric protein member of a family of enzymes known to have several active quaternary structures in different species (monomers, homodimers, homohexamers, and possibly others). These enzymes do not appear to suffer the large conformational transitions observed in many allosteric enzymes, and have a single active site per monomer, formed by amino acids clearly distinct from those at the dimer interface (Oksanen et al., 2007).
In our model, a residue was defined as a member of the interface if it had at least one non-hydrogen atom within 3.7 Å of the opposite subunit. Choosing this short distance restricted the number of residues to those establishing close contacts with the opposing subunit and most likely to produce important energy changes upon mutation. Under this criterion, we were able to identify 22 residues belonging to the interface and each of them was independently mutated to alanine (a subtle mutation) or a new residue with properties differing significantly from those of the native residue (an aggressive mutation). All of these, led to a total of 44 different in silico mutation possibilities.
Our simulation protocol can be summarized as follows: Initially, the original PDB file was modified to add all hydrogen and missing atoms. Then, as a starting point, equilibrium structures were obtained with HyperchemTM (version 7.5 Hypercube, Inc., Gainesville, FL, USA, using Amber 99 force field), and with the Rosetta (Dantas et al., 2003) “idealize” option. We chose two different approaches towards the construction of the mutant structures: The first one provides only one final structure for each mutation, but allows continuous variation of bond-lengths, bond angles, dihedrals, and so on. The second one provides a collection of possible conformers for each structure, but is restricted to the set of rotamers included in the Rosetta libraries.
In our first approach, the 44 mutants were constructed using the HyperchemTM mutation algorithm and their respective equilibrium structures were obtained. A position restrained minimization with fixed backbone atoms was followed by a free minimization in vacuum. Clearly this procedure is not completely realistic, because it overlooks the role of the solvent, pH, temperature, and possible effects of the mutations on the folding pathway. Nevertheless, focusing in one aspect at a time may be a desirable feature, since the negative impact of these limitations can be reduced if, at some point, the teacher points out to the students the pro et contra of the simulations.
With Rosetta, the approach was to design the mutant using the fixed backbone option, and taking at least ten different solutions for each mutant. This approach has very much the same limitations mentioned above, but renders a collection of side-chain conformers for each mutant, useful to add some impression of movement to the otherwise frozen structure. Using full molecular dynamics simulations is another possibility worthy of future consideration, but this procedure is time-consuming for a large set of mutations, and yields very large data files for each result.
VisualizationVisualization of the structures was achieved with the aid of VMD (Humphrey et al., 1996). This program was selected because it has the following features:
- i)
An integrated, general-purpose, and well documented computer language interpreter (Tcl/Tk). Tcl/Tk has been successfully used to create large applications with friendly graphical user interfaces (Welch et al., 2003).
- ii)
Real-time traceable variables capturing the interactions between the user and the displayed molecule.
- iii)
High quality graphics with several 3D stereo modes suitable for most virtual reality facilities available.
- iv)
Free for academic use, which makes it available to advanced and motivated students for its use after the IXTLI experience.
- v)
Comprehensive documentation.
- vi)
Highly customizable, and able to read many molecular file formats.
- vii)
Capable to deal with large files, multiple structures, and follow molecular-dynamics trajectories.
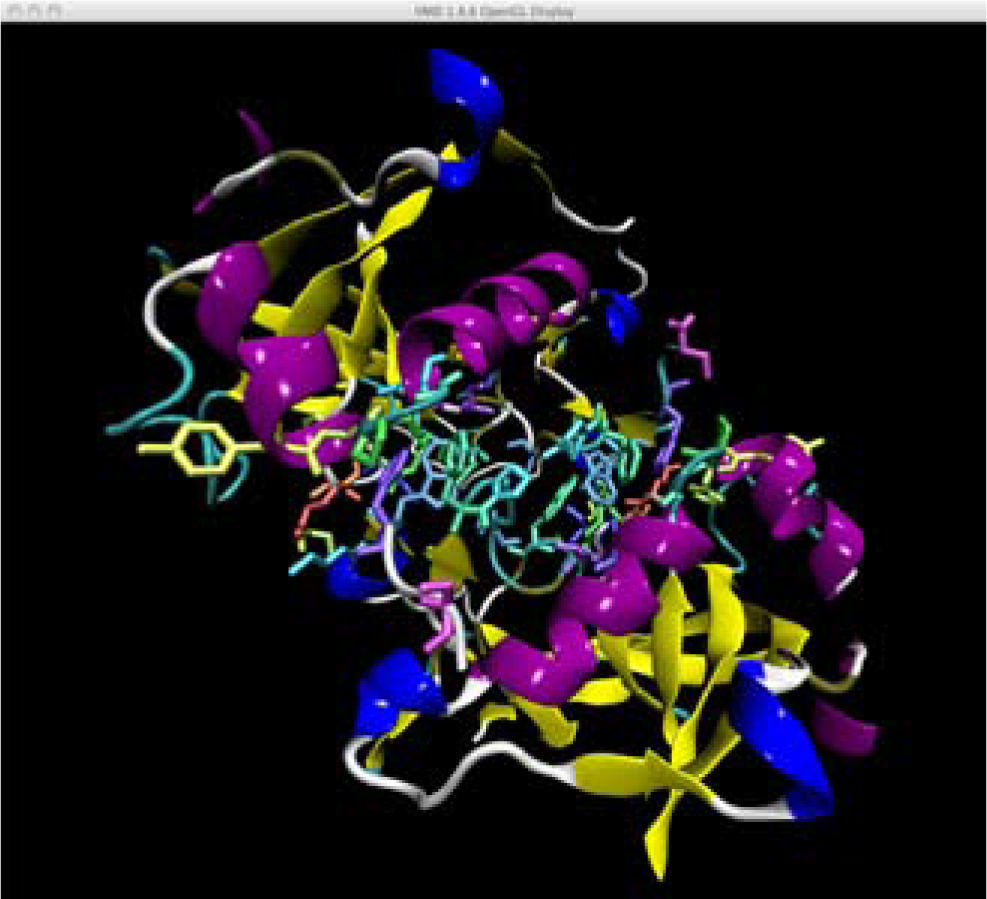
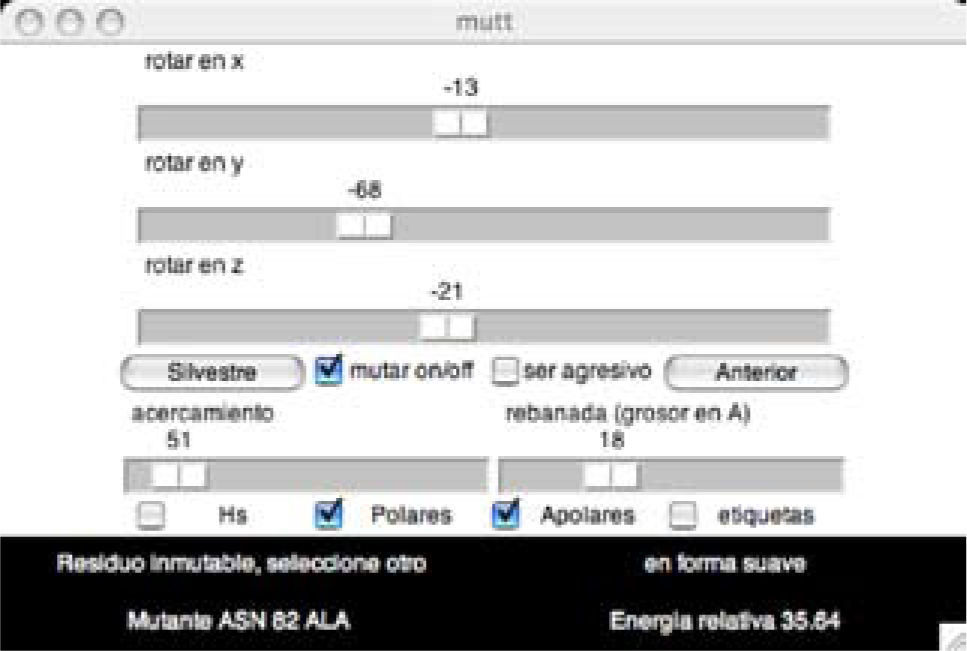
The user interface includes the 3D protein structure image generated on an OpenGL VMD window (Figure 1), and a user control widget (Figure 2) written in Tcl/Tk 8.4 directly linked to VMD commands. It can be downloaded from internet, together with the mutant database (http://depa.pquim. unam.mx/proteinas/ixtli/index.html). At the same site, additonal information is available regarding the pyrophosphatases, and the methods employed are described in more detail. The user interface has the following features:
- i)
It automatically sets the environment, loads the native enzyme file, and sets the backbone cartoon view, and only the side chains of interface residues are shown as “licorices”.
- ii)
A slider bar is used to move two clipping planes parallel to the dimer interface to control the displayed elements from the full view to a narrow slice of the interface. These clipping planes are always maintained parallel to the interface plane (i.e. they move along with the structure), so this feature is not equal to a conventional Z-slab such as can be found in SwissPDBviewer (Guex and Peitsch, 1997).
- iii)
A second set of sliders allows individual rotations of the X, Z or Y axis, or zooming in and out. This feature is redundant to VMD native controls, but it prevents inexperienced users from moving the molecule out of the scene.
- iv)
Controls to toggle the display of hydrogen atoms, residue labels, and polar or non-polar amino acids at the interface.
- v)
Buttons to return to the native enzyme structure and to recall the previous selected mutant.
- vi)
Controls to toggle the mutation mode on and off, and to select from the subtle, or the aggressive mutant sets.
- vii)
Tracing of the picked atoms (those clicked with the pointer) when the mutation mode is on. Under this condition, the program records the molecule orientation, and displayed state, loads the corresponding file, and displays it with the same representation, perspective and orientation as the previously loaded. The operations give the impression of an “instant mutation” taking place on the displayed molecule.
- viii)
Text field to indicate the mutant name, its energy change relative to the native protein, and essential status information.
- ix)
The program call can be made from the command line, so it can be reduced to a single command. Command line arguments control the state of the stereographic projection mode, so the software can also be run in a small computer without any special hardware. This feature is especially useful to become familiar with the program, and to test new teaching protocols.
, but it can be changed at will from 5 (only the interface side chains are shown) to 50 Å (full view).")
OpenGL window of the protein under display. Notice how the backbone cartoons have been sliced both at the front and the back of the structure. The slice width is ± 18 Å from the interface (as indicated by the widget in Figure 2), but it can be changed at will from 5 (only the interface side chains are shown) to 50 Å (full view).
 and last selected mutant (“Anterior”). The upper check boxes toggle the mutation mode on (“mutar on/off”), and the aggressiveness level of the mutation to be performed, which can be subtle (for alanine), or aggressive if the “ser agresivo” box is checked. The lower checkboxes toggle the display of hydrogen atoms (Hs), polar (“Polares”) or non-polar (“Apolares”) interface side chains as well as the labels (“etiquetas”). The text box indicates the mutation status of the selected amino acid, the mutant displayed and the energy relative to the wild type (in amber force field units).")
User control widget. The three upper sliders control rotation. The lower sliders control the zoom and the width of the visible slice around the dimer interface. The central buttons display the wild type (“Silvestre”) and last selected mutant (“Anterior”). The upper check boxes toggle the mutation mode on (“mutar on/off”), and the aggressiveness level of the mutation to be performed, which can be subtle (for alanine), or aggressive if the “ser agresivo” box is checked. The lower checkboxes toggle the display of hydrogen atoms (Hs), polar (“Polares”) or non-polar (“Apolares”) interface side chains as well as the labels (“etiquetas”). The text box indicates the mutation status of the selected amino acid, the mutant displayed and the energy relative to the wild type (in amber force field units).
To actually test our interface in a class session, 16 chemistry undergraduate students enrolled on the 7th term (out of 9) were invited to the session. It is important to mention that these students already had a course in Cell Biochemistry in the 6th term. They took conventional classes on protein structure, with images of 3D structure representations from textbooks. Some mentioned having some experience with interactive 3D visualization programs, but none of them had attended before a session at the Virtual Reality Observatory ixtli-unam.
The session started with a brief description of the enzyme under study and a quick guide to the program interface. During this introduction, short help notes were handed out to them.
Following the introduction, the students were separated in four teams and given an assignment sheet (Figure 3). They were given 5 minutes in order to get used to the relevant visual aspects of the program and then were given 10 more minutes to tackle the assignment using the software. The session was video recorded for later analysis of the students' performance.
.")
While working, they were asked to discuss their work aloud, so their classmates and the teachers could hear their thoughts. The idea here was to allow the teams to learn from each other. At the end, in addition to their answers to the assignment, they were asked to give their personal impressions on the software as a learning tool, to judge their own performance, and to answer a small questionnaire. After the session, the teachers discussed the group findings and offered some guidance.
ObservationsTeam # 1This team was highly cooperative during their work session; nevertheless two of its members took the leadership and drove their teammates to the final decisions. They showed a marked tendency to select aggressive mutations and were very surprised by the non-local changes in the protein structure caused by the mutations. They also asked questions to the rest of the group.
Team # 2In this team, one of its members directed all the work done, their selections were systematic and showed more insight into the protein structural changes brought about by the mutations. They paid more attention to the energy values after every mutation and examined changes in residue contacts in the protein. For example, several times after a mutation, they went back to the wild type and forth in their attempt to correlate energy values with structural changes.
Team # 3This team was the less skillful in their approach and showed a poorer understanding of the mutation results. The amino acids to be mutated were chosen almost at random and the team members chose only non-aggressive changes. Once the resulting protein structure appeared, they paid attention to the protein general view and focused on the properties of the chosen amino acid. It was very difficult for them to find one amino acid that would give a large energy change. At some point, they also turned to the other teams to get some ideas.
Team # 4Members of this team were very surprised and intrigued by the results they obtained with the different mutations. Whenever they obtained the mutated protein structure, they were primarily interested in the protein general shape and secondarily they looked for repulsive interactions between the newly introduced amino acid and the other protein residues.
For this team, it took less time to adapt themselves to the three-dimensional aspects of the protein, as they benefited from watching the other teams at work.
In general, students with a better knowledge of the subject were clearly more confident and asked fewer questions regarding the use of the software. Some of them even showed well-developed skills in the handling of the 3D molecular representations. When questioned about their previous experience, this advanced group of students reported previous extensive contact with molecular visualization software. Nevertheless, all of them considered the use of this software in the virtual reality environment a very stimulating and complete experience, because they felt able “to travel inside the protein guts”, and see factors they had not previously appreciated in two-dimensional representations of proteins. Interestingly, in their comments, all of them made reference to an improved understanding of the interactions between amino acids, their role in protein structural changes, and their relationship with the overall stability.
Afterwards, many of the students registered for a “General session” which is offered on a regular basis by IXTLI-UNAM, where virtual reality software for architecture, earth science and other areas is demonstrated to visitors.
In addition to the general comments on the students' performance, students with less understanding of the subject, or with less experience in the use of molecular visualization software, required some guidance to gain enough confidence with the interface. Nevertheless, development of basic skills took them from 2 to 5 minutes. We considered this as an indication of the interface being reasonably user-friendly.
Demonstration to Biochemistry lecturersIn addition to the above session with students, the software was also demonstrated to invited lecturers and professors from the Chemistry Faculty at UNAM. All of them teach biochemistry at the undergraduate level, and some teach it at the postgraduate level too. After an explanation of the software features and limitations, we invited them to come down and use the software. Surprisingly, and contrary to the students, most of them were reluctant to take the controls. Instead, they raised several points, mainly related to improvements of the visual effects of the program, or the addition of extensive features to the software, like enzyme activity changes and hydrogen bonding patterns. Only two, out of the twenty present, actually came forward to manipulate the images personally.
At the end of the session, we asked the participants to give their impressions and suggestions in written form. One of the participants refused to give his view, but the remaining twelve opinions were favorable, and found the software a useful teaching aid. Many suggested changes to increase the complexity of the interface and its capabilities.
Final commentThe effects of mutations in proteins can be a powerful teaching-aid in a classroom. But the present computer systems and our knowledge of protein three-dimensional structure imposes tight limits to the development of flexible software. Yet, as shown here, useful applications can be developed, if their scope is restricted to a specific problem.
Interestingly, students were eager to use the software presented here, while teachers were reluctant. On the other side, students liked the user interface simple, as it was; while teachers requested the inclusion of several, more complex capabilities. The reason behind those differences may deserve further consideration.