





21-hydroxylase deficiency (21-OHD) is the most common form of congenital adrenal hyperplasia (CAH). In adulthood, most studies are reported in females. By contrast, data on adult males are scarce.
ObjectiveTo describe a series of adult males with classic 21-OHD and to assess the presence of adrenal masses and testicular adrenal rest tumors (TARTs).
Material and MethodsEight males (21–42 years) were included. We evaluated clinical presentation, 17-Hydroxyprogesterone (17-OHP), Testosterone (T), Δ4Androstenedione (Δ4A) ACTH, LH, FSH and plasma renin activitiy (PRA) levels at consultation. Molecular studies of the CYP21A2 gene, testicular ultrasound (US), semen analysis and adrenal computed tomography (CT) scan were performed. Treatment and compliance were assessed.
ResultsBasal 17-OHP levels were >20ng/ml in all patients. At consultation, median 17OH-P was 11.5 (2.3–81) ng/ml, FSH: 3 (0.3–4) mUI/ml, LH: 1.1 (0.1–6) mUI/ml, T: 4.3 (1.7–8) ng/ml, Δ4A: 5.7 (1.4–16) ng/ml, ACTH: 86.4 (76–334) pg/ml, PRA: 9.5 (1.3–23.6) ng/ml/h. Semen analysis was performed in 5/8 patients, showing azoospermia in two. Molecular genetic analysis was performed in 4/8 patients. TARTs were found in 5/6, being bilateral in four. Adrenal masses were found in 4/6. In the 7 patients diagnosed in childhood, their follow-up was referred to as irregular, both in their attendance at consultations and in compliance with the indicated treatment.
ConclusionsTo our knowledge, this is the first series on adult males with classic 21-OHD which concomitantly assesses clinical presentation, molecular biology, adrenal and testicular imaging studies, semen analysis and compliance to treatment. A high prevalence of adrenal masses and TARTs was observed, possibly associated with poor treatment compliance leading to elevated ACTH and increased proliferation. Our findings on TARTs agree with reports in international publications of CAH in males, with adrenal imaging being added in our group. Although we are aware that further studies with a larger sample size and more data are needed, we consider that our findings contribute to the clinical management of classical 21-OHD in the male population.
El déficit de 21-hidroxilasa (21-OHD) es la forma más frecuente de hiperplasia adrenal congénita (HAC). La mayoría de las publicaciones en adultos se refieren a la población femenina, habitualmente, los reportes en varones adultos son escasos.
ObjetivoDescribir una serie de varones adultos con 21-OHD clásica y determinar en ellos la presencia de masas adrenales y restos adrenales testiculares (TART).
Materiales y métodosSe incluyeron 8 varones adultos de entre 21 y 42 años de edad. Se evaluó la presentación clínica, 17-hidroxiprogesterona (17-OHP), testosterona (T), Δ4Androstenediona(Δ4A) ACTH, LH, FSH y actividad de renina plasmática (ARP). Se solicitaron: genética molecular del gen CYP21A2, realizándose en 4/8. Se efectuó ecografía testicular, espermograma y TAC de suprarrenales. Se consignó tratamiento indicado y su cumplimiento.
ResultadosLa 17OHP basal fue mayor a 20ng/ml en todos los pacientes Los valores hormonales a la consulta fueron: 17-OHP: 11,5 (2,3-81) ng/ml, FSH: 3 (0,3-4) mUI/ml, LH: 1,1 (0,1-6) mUI/ml, T total: 4,3 (1,7-8) ng/ml, Δ4A 5,7 (1,4-16) ng/ml, ACTH 86,4 (76-334) pg/ml, ARP 9,5 (1,3-23,6) ng/ml/h. Se efectuó espermograma en 5/8, hallándose azoospermia en 2. Se detectaron TART en 5/6, siendo bilaterales 4. Se hallaron masas adrenales en 4/6. En los 7 pacientes diagnosticados en la infancia, su seguimiento estaba referido como irregular tanto en la concurrencia a las consultas como en el cumplimiento del tratamiento indicado.
ConclusionesHasta donde sabemos, esta es la primera serie en varones adultos con 21-OHD clásico que evalúa concomitantemente la presentación clínica, la biología molecular, los estudios de imágenes suprarrenales y testiculares, el análisis de semen y el cumplimiento del tratamiento. Se observó una alta prevalencia de masas suprarrenales y TART, posiblemente asociada con un cumplimiento deficiente del tratamiento que conduce a una ACTH elevada y mayor proliferación. Nuestros hallazgos sobre TART están de acuerdo con los informes en publicaciones internacionales de HAC en varones, agregando imágenes suprarrenales concomitantes en nuestro grupo. Aunque reconocemos que se requieren más estudios con un mayor tamaño de muestra y de datos, consideramos que nuestros hallazgos contribuyen al manejo clínico del 21-OHD clásica en la población masculina.
Congenital adrenal hyperplasia (CAH) comprises a group of inherited autosomal recessive disorders characterized by an enzymatic defect in adrenal cortisol biosynthesis.1,2 Deficiency of the 21-hydroxylase (21OHD) enzyme is the most common cause of CAH, accounting for approximately 95% of the cases. The deficiency is caused by mutations in the CYP21A2 gene located on the short arm of chromosome 6 leading to a decrease in cortisol biosynthesis and increased ACTH, 17-hydroxyprogesterone (17-OHP), and adrenal androgen levels. According to the severity of the enzyme deficiency, two phenotypes are defined: the classical CAH and the non-classical forms. Classical CAH has an incidence of 1/12,000–1/20,000 in newborn infants, with two different clinical presentations: the more severe salt-wasting (SW) type with almost no enzyme activity and the simple virilizing (SV) type, with an enzyme activity of 3–7%. The SW type presents with adrenal crises in the neonatal period and virilization3–5 and the SV form with virilization and generally no adrenal crises.
The majority of studies on classical 21OHD are reported in females. On the other hand, reports of 21OHD in adult males are scarce: the diagnosis is generally made in the context of genetic studies requested because of an affected member of the familiy or due to a perinatal adrenal crisis. In fact, the majority of the bibliographic reports on CAH in males describe older children and adolescents.2,6,7
As treatment with glucocorticoids has only been available since the last six decades, it is not common to find patients older than 60 years with 21OHD that suffer from the long-term consequences of this disorder.1–4 Treatment with glucocorticoids (and mineralocorticoids when needed) reduces the risk of adrenal crises and androgen excess, controlling the disease symptoms. The presence of adrenal masses and testicular adrenal rest tumors (TARTs) has been described in 21OHD, predominantly in inadequately treated patients.
The aim of our study is to describe a series of classical 21-OHD adult males studied at our Endocrinology Department in order to assess the following parameters: clinical features, hormonal profile, molecular studies, semen analysis and the presence of TARTs and adrenal masses.
Material and methodsPatientsWe studied 8 adult male patients with classical 21OHD: seven subjects were referred to our hospital for follow-up of 21-OHD CAH diagnosed in their infancy. The eighth patient consulted us in adulthood due to an adrenal incidentaloma and whose diagnosis of 21-OHD was assessed while performing hormonal studies. We would like to point out that at the Endocrinology Department of our Hospital, we study adult CAH patients and we perform the follow up of those CAH patients originally diagnosed and treated at pediatric centers.
This is a retrospective transversal study of adult patients, over 18 years-old, who were attended at our hospital between January 2000 and June 2017. For phenotypic classification of the patients, we recorded the clinical data, the personal history of adrenal crises in childhood, glucocorticoid and mineralocorticoid treatment (type, dose and compliance). Compliance was established by assessing regular attendance to clinical control, uninterrupted treatment and laboratory determinations in the treatment goals (see “Laboratory Studies”). Weight, height, body mass index (BMI), blood pressure, presence or absence of cushingoid stigmata and urogenital examination were assessed.
Routine laboratory studies, hormonal profile, molecular studies of CYP21A2 gene, semen analysis, testicular ultrasound and non-contrast adrenal computed tomography (CT) scan were performed.
Laboratory studiesHormonal profilePlasma ACTH reference range (RR): 0–46pg/ml), FSH (RR: 1.5–12.4mUI/ml), LH (RR: 1.7–8.6mUI/ml), and total testosterone (T) (RR: 2.7–8.2ng/ml) were determined by electrochemiluminescence assays. Bioavailable T was calculated by Vermeulen formula (sex hormone binding globulin -SHBG).8,9 Δ4Androstenedione (Δ4A) (RR: 0.62–3.12ng/ml) and 17-OHP (RR: 0.6–3.4ng/ml) were measured by radioimmunoassay (RIA). Plasma renin activity (PRA) was measured using solid-phase RIA Diasorin, Beckman normal range supine: 0.79 (0.34–1.9) ng/mL/h.
As for patients under treatment, Δ4A and total T levels in the mid-to-upper range were considered as appropriate for treatment goals.
Routine laboratory studiesGlucose levels, plasma ionogram, cholesterol (total, low-density lipoprotein, and high-density lipoprotein) and triglycerides were measured between 8–9.30 a.m. after a 12-h fast.
Laboratory data collected at the first visit to our Department was used for evaluation.
Molecular study of the CYP21A2 geneMolecular studies were performed at the Molecular Biology Laboratory of the Department of Endocrinology of Garrahan Hospital, Buenos Aires, Argentina. The 11 most common mutations (p.Pro30Leu: g.89C>T, c.89C>T; In2: g.656C>G, c.293-13C>G; Del8bpE3: p.Gly110ValfsTer21, g.708_715delGAGACTAC, c.329_336delGAGACTAC; p.Ile172Asn: g.1000T>A, c.515T>A; ClEx6: p.[Ile236Asn;Val237Glu;Met239Lys]; g.[1382T>A; 1385T>A; 1391T>A], c.[707T>A; 710T>A; 716T>A], p.Val281Leu: g.1685G>T, c.841G>T; p.Gln318Ter: g.1996C>T, c.952C>T; p.Arg356Trp: g.2110C>T, c.1066C>T; p.Pro453Ser: g.2581C>T, c.1357C>T; p.Arg483ProfsTer58: g.2671_2672delGGinsC, c.1447-1448delGGinsC; Del/Conv: large rearrangements (30-kb deletion including 3’ of CYP21A1P and 5’ of CYP21A2 and CYP21A2 converted to CYP21A1P in the 5’ part) were studied using DNA from leukocytes in peripheral blood obtained according to standard methods.10
NCBI Reference Sequence: NG_007941.3. Numbering starts from A in the initiation codon
Testicular examinationIn all patients, testicular examination was performed by palpation and testicular volume was measured using a Prader orchidometer. Additionally, testicular ultrasonography was performed using a soft-tissue transducer (7–15MHz).
Semen analysisSemen analysis was performed at the Male Fertility Laboratory of our hospital. Semen samples were obtained after a sexual abstinence of 3 to 5 days and analyzed according to the WHO 2010 criteria.11
Fertility potential was analyzed according age at diagnosis, clinical features, and adherence to treatment.
Abdominal computed tomography (CT) scanNon contrast abdominal CT scan was performed according to the protocol for adrenal glands, including thin slice images to determine size, aspect and lipid content expressed by density in Hounsfield Units (HU).
Statistical analysisIBM SPSS (Statistical Program for Social Sciences) software version 21 was used for statistical analysis.
ResultsEight adult males with 21OHD were included in the study. Mean age was 25.6±7.3 years. Three patients had the SW form and five the SV form. In the three SW patients, the diagnosis was made during the first month of life due to an adrenal crisis. In the SV patients, the diagnosis was established during infancy in four: two were studied due to a family history (affected sisters); one presented an adrenal crisis in the first month of life and the fourth patient had precocious pubarche when he was 4 years-old. The remaining SV patient (patient 8) was a 28-years-old male who consulted us because of an adrenal incidentaloma found in an abdominal sonographic scan which was performed due to a gallbladder lithiasis; an adrenal CT scan confirmed the adrenal mass and his hormonal profile led us to the diagnosis of classical 21-OHD- full description of the patient is under the “adrenal masses” section.
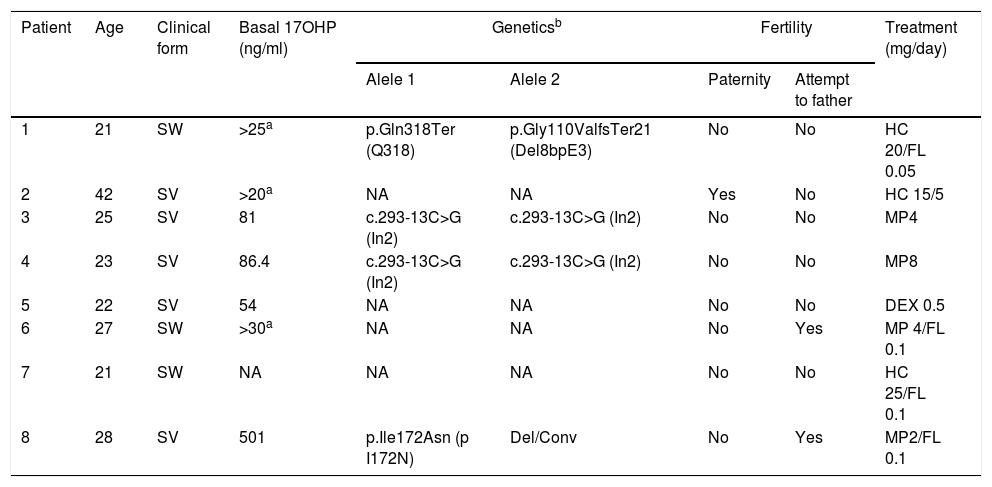
Clinical data, classical 21-OHD forms, basal 17OHP, levels, molecular genetic studies, fertility status and treatment are listed for each patient in Table 1.
Clinical forms, basal 17OHP levels, molecular genetic studies, fertility and treatment in men with classical 21-OHD CAH*.
| Patient | Age | Clinical form | Basal 17OHP (ng/ml) | Geneticsb | Fertility | Treatment (mg/day) | ||
|---|---|---|---|---|---|---|---|---|
| Alele 1 | Alele 2 | Paternity | Attempt to father | |||||
| 1 | 21 | SW | >25a | p.Gln318Ter (Q318) | p.Gly110ValfsTer21 (Del8bpE3) | No | No | HC 20/FL 0.05 |
| 2 | 42 | SV | >20a | NA | NA | Yes | No | HC 15/5 |
| 3 | 25 | SV | 81 | c.293-13C>G (In2) | c.293-13C>G (In2) | No | No | MP4 |
| 4 | 23 | SV | 86.4 | c.293-13C>G (In2) | c.293-13C>G (In2) | No | No | MP8 |
| 5 | 22 | SV | 54 | NA | NA | No | No | DEX 0.5 |
| 6 | 27 | SW | >30a | NA | NA | No | Yes | MP 4/FL 0.1 |
| 7 | 21 | SW | NA | NA | NA | No | No | HC 25/FL 0.1 |
| 8 | 28 | SV | 501 | p.Ile172Asn (p I172N) | Del/Conv | No | Yes | MP2/FL 0.1 |
classical 21-OHD CAH - OMIM 201910 (nomenclature).
Nomenclature for different mutations at the protein and nucleotide level is the following: p.P30L: g.89C>T, c.89C>T; In2: IVS2-13A/C>G, g.656A/C>G; Del 8bp E3: p.G110VfsX21, g.708_715delGAGACTAC, c.329_336delGAGACTAC; p.I172N: g 1000T>A, c.515T>A; ClEx6:
p.[I236N;V237E;M239K]; g.[1382T>A; 1385T>A; 1391T>A], c.[707T>A;710T>A;716T>A], p.V281L: g.1685G>T, c.841G>T; p.Q318X: g.1996C>T, c.952C>T; p.R356W: g.2110C>T, c.1066C>T; p.P453S: g.2581C>T, c.1357C>T; p.R483PfsX58: g.2671_2672delGGinsC, c.1447-1448delGGinsC; DEL/CONV: large rearrangements (a 30-kb deletion including the 3’ region of the CYP21A1P and the 5’ region of the CYP21A2 gene and macroconversions of the CYP21A1P pseudogene that are converted to the CYP21A2 gene). NCBI Reference Sequence: NG_007941.2. Numbering starts at the A of the first ATG.
SW: salt-wasting type; SV: simple virilizing type HC: hydrocortisone; FL: Fludrocortisone; MP: methylprednisolone; Dex: dexamethasone.
NA: Not analyzed
Mean height was 1.62±0.1m, mean BMI was 24.7±4.5kg/m2, mean systolic blood pressure (SBP) was 106±5.2mmHg, and mean diastolic blood pressure (DBP) was 62.5±7.1mmHg. No significant differences were found between the two groups,
One patient was obese (SV), three were overweight (2 SW and 1 SV). None of them had cushingoid features. Four patients had a normal weight (3 SV and 1 SW). None of the patients had arterial hypertension.
Glucocorticoid replacement therapy: The most frequently used glucocorticoids were methylprednisolone and hydrocortisone. Four patients received fludrocortisone (Table 1).
It is important to point out that the 7 patients with previous diagnosis showed irregular adherence to treatment since childhood.
Laboratory studiesHormonal determinations: Patients 1–7 were already treated and patient 8 was recently diagnosed and had not begun treatment at the time of hormonal studies. Individual data of basal 17OH-P levels (available in 7/8) is shown in Table 1. In patients 1–7, basal 17OHP values correspond to the determinations of the original laboratory at the time of diagnosis in infancy. Median 17OH-P levels in patients who were under treatment was 11.5 (2.3–81) ng/ml; in patient 8, recently diagnosed at our hospital, basal 17OH-P levels were 501ng/ml. Median gonadotropin (Gn) levels were: LH: 1.1 (0.1–6) mUI/ml and FSH: 3 (0.3–4) mUI/ml.
Median total T was 4.3 (1.7–8) ng/ml. Bioavailable T levels were available for 5/8 patients with a median of 1.35 (0.83–1.6) ng/ml. Δ4A: was determined in eight patients and the median was 5.7(1.4–16). ng/ml. Data for ACTH and PRA were available in 5/8 and in 4/8 patients, respectively – with a median of 86.4pg/ml (76–334) for ACTH and 9.5ng/ml/h (1.3–23.6) for PRA. (not shown).
Routine laboratory studies: Mean glucose levels were 87±13mg/dl; total cholesterol was 228±116mg/dl; LDL:155±97mg/dl; triglycerides:139±147mg/dl; sodium:137±3mEq/l; potassium:4.3mEq/l.
Molecular genetic studiesOnly four patients perform analysis of the CYP21A2 gene (Table 1).
Testicular examinationAll patients underwent testicular examination. Two patients (P1 and P2) had testicular hypotrophy (testicular volume ≤15cm3) and additionally one patient had a palpable tumor.
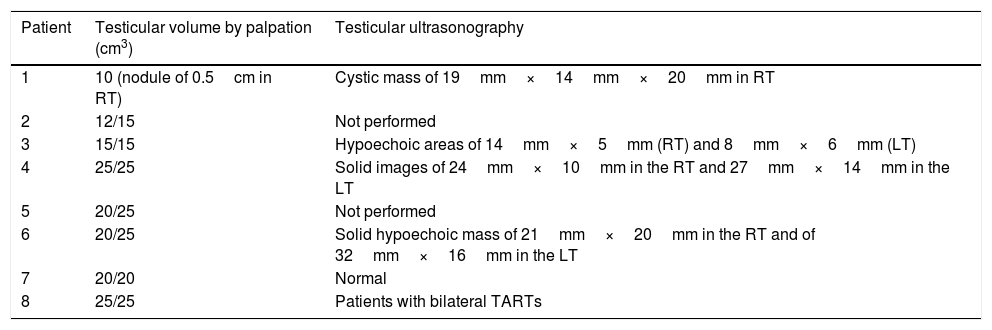
In 6/8 patients, testicular ultrasonography was performed. TARTs were identified in 5/6 and they were bilateral in 4/5. Patient 4 required a testicular biopsy due to TARTs growth (Table 2).
Testicular evaluation.
| Patient | Testicular volume by palpation (cm3) | Testicular ultrasonography |
|---|---|---|
| 1 | 10 (nodule of 0.5cm in RT) | Cystic mass of 19mm×14mm×20mm in RT |
| 2 | 12/15 | Not performed |
| 3 | 15/15 | Hypoechoic areas of 14mm×5mm (RT) and 8mm×6mm (LT) |
| 4 | 25/25 | Solid images of 24mm×10mm in the RT and 27mm×14mm in the LT |
| 5 | 20/25 | Not performed |
| 6 | 20/25 | Solid hypoechoic mass of 21mm×20mm in the RT and of 32mm×16mm in the LT |
| 7 | 20/20 | Normal |
| 8 | 25/25 | Patients with bilateral TARTs |
RT: right testis; LT: left testis.
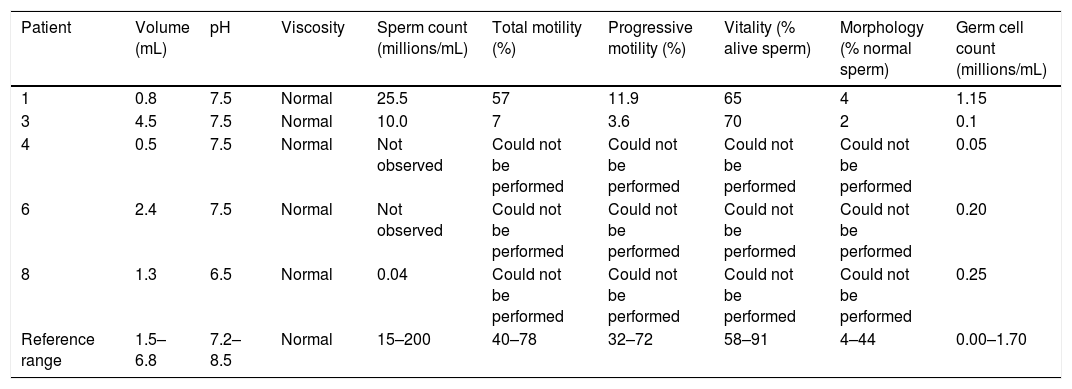
Semen analysis was performed in five patients. Azoospermia was observed in two. The results are shown in Table 3.
Semen analysis.
| Patient | Volume (mL) | pH | Viscosity | Sperm count (millions/mL) | Total motility (%) | Progressive motility (%) | Vitality (% alive sperm) | Morphology (% normal sperm) | Germ cell count (millions/mL) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.8 | 7.5 | Normal | 25.5 | 57 | 11.9 | 65 | 4 | 1.15 |
| 3 | 4.5 | 7.5 | Normal | 10.0 | 7 | 3.6 | 70 | 2 | 0.1 |
| 4 | 0.5 | 7.5 | Normal | Not observed | Could not be performed | Could not be performed | Could not be performed | Could not be performed | 0.05 |
| 6 | 2.4 | 7.5 | Normal | Not observed | Could not be performed | Could not be performed | Could not be performed | Could not be performed | 0.20 |
| 8 | 1.3 | 6.5 | Normal | 0.04 | Could not be performed | Could not be performed | Could not be performed | Could not be performed | 0.25 |
| Reference range | 1.5–6.8 | 7.2–8.5 | Normal | 15–200 | 40–78 | 32–72 | 58–91 | 4–44 | 0.00–1.70 |
* Reference values correspond to the 5–95% range according to the WHO 2010.
In five patients, non-contrast abdominal CT scan was performed according to the protocol for adrenal glands and nodules were identified in four. The features are shown in Table 4.
Adrenal CT scan.
| Patient | CT scan |
|---|---|
| 1 | Pseudonodular image of 6mm×12mm, 13 HU in the RAG |
| 2 | Bilateral thickening with left predominance of less than 13mm, compatible with myelolipomas |
| 3 | Normal |
| 4 | Nodular image of 15mm×17mm in the LAG |
| 8 | Nodular image of 49mm×34mm 32 HU in the RAG. |
RAG: right adrenal gland; LAG: left adrenal gland; HU: Hounsfield units.
Two of the cases warrant additional comments:
Patient 4: The patient consulted at 23 years of age for the follow-up of 21OHD-SV diagnosed at 3 months of life because of an affected sister. He received treatment with hydrocortisone, with irregular adherence during childhood. At 18 years of age, bilateral TARTs were found on testicular ultrasonography. Hydrocortisone was switched to methylprednisolone and bilateral testicular biopsy was performed due to TARTs growth. In both biopsy specimens, Leydig-cell hyperplasia was observed with positive immunostaining for inhibin; and absence of sperm motility was found.
Patient 8: The patient consulted at our department when he was 28 years-old because of a right adrenal solid mass, 49mm×34mm and 32HU found during the workup for symptoms of biliary colic. Due to the mass characteristics, 17OHP was requested and it was 501ng/ml. Molecular study of the CYP21A2 gene confirmed the diagnosis of 21OHD –SV form–, Allele 1: p .Ile172Asn and Allele 2 DEL/CONV. Due to the size and suspicious aspect of the adrenal mass, surgery was performed: and the histological diagnosis was a cortical adrenal adenoma. He has no children at the moment, sperm cryopreservation has been performed and he is seeking fertility. We would like to remark that he had no previous symptoms that could have led to CAH diagnosis and that he had never consulted an endocrinologist before.
DiscussionWe describe eight adult male patients with classical 21OHD, assessing the clinical, laboratory, and molecular features as well as the presence of adrenal masses, TARTs, and semen analysis. Although the series is small, the sample size is in agreement with other reports in the literature in which only a few patients were recruited.3,12–15 There is, however, an extensive and detailed series by Bouvattier et al.,5 but without data about concomitant adrenal imaging studies.
In our country and for decades, boys with 21OHD were only diagnosed in urban centers and many of them died because of dehydration in the first hours of life. Indeed, the detection of SW forms may continue to be underdiagnosed. In Argentina, the neonatal screening program law was implemented in 200716 with a CAH incidence of 1:8937 (female to male ratio 0.8) in Buenos Aires.17 Patient 8, who had no previous diagnosis, was born in a neighboring country where there was no neonatal screening performed at the time of birth.
Regarding the clinical features, none of the patients in our series were hypertensive, in agreement with the majority of the studies in the literature.3,5 This suggests that probably our patients were not overtreated; indeed, the majority of them had irregular compliance to treatment. It is noteworthy, that in the study by Finkielstain et al.18 in a series of patients with CAH of both sexes, those with classic CAH had a higher rate of hypertension than those with non-classic CAH.
All the patients in our series, except patient 8 with a recent diagnosis, had been receiving glucocorticoids. Dexamethasone was the least commonly used, in agreement with reports from France and the United Kingdom. Patient 8 (SV), is receiving methylprednisolone and fludrocortisone, in order to achieve a better control and to avoid a larger glucocorticoid dose along with its possible detrimental effects.
As for the hormonal profile at consultation, only two patients had normal 17OHP levels and one (patient 5) had normal ACTH levels. In the remaining patients these levels were high. It is well known that normalization of 17OHP levels is not a treatment goal since it may result in glucocorticoid overtreatment.1,3
In women, treatment success is assessed by measuring Δ4A and T levels. In men, however, T is not a reliable parameter since it shows gonadal function rather than adrenal one.3 Therefore, in males Δ4A levels are preferably used and they should be maintained at values that are appropriate for age and sex.4 In our series, Δ4A levels were in the treatment goal in only 3 patients and they were high in the remaining ones, suggesting insufficient treatment. Gn levels were normal in four patients, inhibited in three, and FSH was elevated in one (patient 4), probably pointing to a primary testicular failure (data not shown).
Molecular study of the CYP21A2 could be performed in only 4/8 patients; it is noteworthy that in our series the SV form is predominant over the SW clinical form- although it is well known that the expected distribution is 75% for the SW and 25% for the SV. We have no real explanation for this fact, but it is possible that some patients could have missed attendance during follow up at the original institution or that they attended other centers (we have no available data concerning this point).
One aim of our study was to assess the presence of TARTs and the fertility potential of our patients based on semen analysis. It is believed that TARTs result from adrenocortical remnants that descend with the testis and grow under ACTH stimulation.12,19 There is an alternative hypothesis that in patients with 21OHD, TARTs originate from an intratesticular totipotent embryonic cell type with features of Leydig as well as adrenocortical cells containing ACTH, angiotensin II, and LH/HCG receptors.20 In our study, five of the six patients that underwent testicular ultrasonography had TARTs. As TARTs were palpable in only one of the patients, testicular ultrasonography showed to be essential for detection of the tumor. Periodic testicular ultrasonography is currently recommended in these patients since adolescence.14,21 In the literature, prevalence of these tumors varies from 0 to 94%,5,14,22–24 depending on the different methods used (palpation, ultrasonography, or magnetic resonance imaging) and the age of the selected patients. Additionally the youngest CAH patient reported with TARTs is a 1.8 year old.25 There was a high rate of bilaterality (4/6) in agreement with the findings by others authors.5,22,24–26 In our study, patients with bilateral and larger TARTs (patients 4 and 6) had azoospermia, suggesting that the presence of the tumor directly affects spermatogenesis. Our findings agree with those reported by Reisch et al.,12 who conclude that the size of the tumor is inversely proportional to the sperm count and motility. TARTs may grow when ACTH levels increase and reduction can be observed after glucocorticoid treatment, improving the fertility potential.18,27,28 However, Engels et al.,24 describe that only 1/16 patient with TARTs reported to be present at age 16 years, showed disappearance under treatment adjustment. Therefore, this matter is still controversial. In the final stages, when the damage is irreversible, therapeutic options are limited.22 Surgery should be considered and gamete preservation proposed, after previous genetic counseling27 as the prognosis is uncertain.24,29,30
In the patients who performed sperm analysis, severe teratozoospermia was found in all of them suggesting that, regardless of the presence of TARTs, the existence of other alterations associated with 21OHD might affects normal sperm production. Patient 4 underwent bilateral testicular biopsy due to TARTs growth. It is noteworthy that he had the highest gonadotropin levels in the series, suggesting possible damage to the normal testicular tissue and obstruction of the seminal tube associated with oligospermatogenesis. TARTs should be differentiated from Leydig cell tumors because both share common histological features. A history of CAH, presence of bilaterality (83% in TARTs vs. 3% in Leydig cell tumors), histology showing Reinke crystalloids, tumor shrinkage in response to glucocorticoid therapy and lack of malignant features have been used to differentiate them.31 Some patients presented hypospermia, which may be caused by hormonal imbalance, retrograde ejaculation or obstructions. Results of different studies show major alterations in the semen analysis and infertility among overweight and obese men.32 In our series, all spermograms were abnormal regardless of the weight. Falhamar et al.22 observed greater impairment of fertility in patients with the I172N genotype (SV), worse metabolic factors and older age. Others authors, suggested that patients with a deletion/conversion have larger TARTs.33 In our series, since molecular genetic studies were performed in only 4 patients, we cannot draw appropriate conclusions about genotype-phenotype correlations.
Regarding adrenal masses, adrenal nodular images were detected in 4/5 patients who performed a CT scan. In patient 2, the image was compatible with myelolipoma. The first case of CAH-associated bilateral adrenal myelolipoma was published in 1997.34 Myelolipoma was detected in 6% of patients with CAH.35,36 Possible etiological mechanisms could be the prolonged stimulation of the adrenal cortex by sustained high ACTH and the presence of embryonic remains of bone marrow in adrenal tissue. Some of them may be very large, requiring removal due to mass effect. Lipoid and hematopoietic elements would migrate to the adrenal stroma in response to unknown factors.37 Collision tumors of 2 or more histologically distinct compounds may be found.38
In patients with classical 21OHD, the incidence of adrenal masses is higher than in the general population; they are usually found under poor disease control, which could also account for larger size tumors.39–41 In 1992, Jaresh et al.42 reported a high incidence of adrenal masses in classical 21OHD patients: 82% in homozygous and 45% in heterozygous cases. The authors suggest that hypersecretion of ACTH, other peptides and proliferative factors play an important role both in the pathogenesis of adrenal hyperplasia and in adrenal tumorogenesis. At our department, adrenal CT scan has been routinely requested in patients with 21OHD since 1993, and we found a high prevalence of adrenal masses in classical CAH. In 26 women with classical CAH, we could perform adrenal CT scan in 14 and adrenal masses were found in 9/14. Four required surgery due to mass characteristics. The histopathology confirmed one adrenal carcinoma, two cortical adenomas and one myelolipoma.43
In conclusion, we believe that the strength of our study relies in assessing together the clinical presentation, hormonal profile, molecular biology, compliance to treatment, semen analysis, testicular and adrenal imaging studies in adult men with classical 21OHD. However, we are aware that there are some limitations to take into account, such as the small sample size, the retrospective nature of the study, and the fact that not all the tests were performed in every patient. Nevertheless, we consider that our study contributes to the clinical approach in the classical 21-OHD adult male population. Prospective studies with a larger sample size and a long-term follow-up are needed to draw further conclusions regarding this disorder.
Conflict of interestThe authors have no interest to disclose.
