




Familial non-medullary thyroid carcinoma (FNMTC) is defined by the presence of 2 or more first-degree family members with differentiated thyroid carcinoma (DTC). The aim of this study is to compare clinicopathological features and prognosis of FNMTC and sporadic carcinoma (SC).
Materials and methodsRetrospective study of DTC included in the hospital database during the period 1990–2018.
ResultsA total of 927 patients were analyzed, 61 of them were FNMTC, with a mean follow-up of 9.7 ± 6.5 years. The prevalence of FNMTC was 6.6%, with a lower TNM staging presentation (P = 0.003) consequence of a higher proportion of tumors smaller than 2 cm (P = 0.003), combined with a greater multifocality (P = 0.034) and papillary histologic subtype (P = 0.022) compared to SC. No significant differences in age at diagnosis (P = 0.347), gender (P = 0.406), neither in other aggressiveness markers (bilaterality, extrathyroidal extension, lymph node involvement and metástasis) were detected. Rate of persistence/recurrence (P = 0.656), disease-free survival (P = 0.929) and mortality caused by the tumor itself (P = 0.666) were comparable. Families with ≥3 affected relatives, had smaller tumors (P = 0.005), more multifocality (P = 0.040) and bilaterality (P = 0.002), as well as a higher proportion of males (P = 0.020). Second generation patients present earlier FNMTC compared to those of the first generation (P = 0.001).
ConclusionIn our study FNMTC presents a lower TNM staging, higher multifocality and papillary variant, with similar aggressiveness and prognosis compared to SC.
El carcinoma familiar de tiroides no medular (FNMTC, familial non-medulary thyroid carcinoma) se define por la presencia de 2 o más miembros familiares de primer grado con carcinoma diferenciado de tiroides (CDT). El objetivo de este estudio es analizar las características clínicas, anatomopatológicas y pronósticas del FNMTC respecto al carcinoma esporádico (CE).
Materiales y métodosEstudio retrospectivo de los casos de CDT registrados en nuestro centro hospitalario durante el periodo 1990–2018.
ResultadosSe analizó a un total de 927 pacientes, de los cuales 61 son FNMTC, con un seguimiento medio de 9,7 ± 6,5 años. La prevalencia del FNMTC es del 6,6%, presentando un estadio TNM inicial más bajo (p = 0,003) debido a la mayor proporción de tumores inferiores a 2 cm (p = 0,003), además de ser más multifocales (p = 0,034) y del subtipo histológico papilar (p = 0,022), respecto al CE. No se detectaron diferencias significativas en la edad al diagnóstico (p = 0,347), género (p = 0,406), ni en otros marcadores de agresividad (bilateralidad, extensión extratiroidea, afectación ganglionar y metástasis). La tasa de persistencia/recurrencia (p = 0,656), supervivencia libre de enfermedad (p = 0,929) y mortalidad ocasionada por el propio tumor (p = 0,666) fueron comparables. En las familias con ≥ 3 familiares afectados, los tumores son más pequeños (p = 0,005), más multifocales (p = 0,040) y bilaterales (p = 0,002), además de haber una mayor proporción de hombres (p = 0,020). Los pacientes de la segunda generación presentaron al diagnóstico el FNMTC a una edad más precoz respecto a los de la primera (p = 0,001).
ConclusiónEl FNMTC presenta en nuestro estudio, en comparación con el CE, una estadificación TNM más baja, mayor multifocalidad y representación del subtipo papilar, con similares datos de agresividad y sin diferencias en el pronóstico.
Thyroid carcinoma (TC) accounts for 1% of all cancers; it is the most common type of endocrine cancer, and its incidence has been growing in recent decades in many countries around the world.1,2 However, its mortality rate has remained stable at 0.6% per year, since most cases are subclinical (papillary carcinomas under 2 cm).3,4 The different subtypes of thyroid cancer are classified based on their histological origin. Over 90% of carcinomas derive from follicular cells and are called differentiated thyroid carcinoma (DTC); this designation encompasses the papillary, follicular and Hürthle cell carcinoma subtypes.5 A minority of cases arise from parafollicular cells, resulting in medullary thyroid carcinoma.
Familial aggregation is present in non-medullary cases of DTC at a rate of approximately 5%–10%.5 Therefore, familial non-medullary thyroid carcinoma (FNMTC) has been defined as well-differentiated carcinoma originating in follicular cells that presents in two or more first-degree relatives, in the absence of other known predisposing factors of a hereditary or environmental nature.6 Multiple histological variants can coexist in a single family. Some more conservative authors, so as not to overestimate prevalence due to chance, have suggested that at least three relatives be affected.7 In this regard, a mathematical analysis calculated that the estimated probability of DTC being sporadic was 62%–69% with two relatives diagnosed and dropped to 6% with three or more affected relatives.8 In this case, the risk of developing DTC in first-degree relatives of patients diagnosed with TC may be more than three times higher.9,10 Ultrasound screening has been found to detect DTC in up to 22.7% of cases in families with three or more affected members and just 4.6% in families with two affected members; hence, it may be beneficial for the former group. To perform this screening, it is important to take into account the phenomenon of anticipation in the second generation, in which the second generation develops the disease at a younger age and has a more advanced form at diagnosis.11
The genetic bases of these syndromes are complex. Some 5% of cases of FNMTC present on the spectrum of hereditary syndromes (Gardner syndrome, Cowden syndrome, Carney complex, DICER1 syndrome or Werner syndrome) and are called syndromic FNMTC.12 The remaining 95% of cases occur in isolation and are called non-syndromic FNMTC. The genetic causes of the latter remain unclear. The current evidence is in favour of polygenic inheritance mechanisms, with a minority of carcinomas due to mutations located in susceptibility genes.13 These patients have been found to have increased telomere instability compared to healthy subjects and patients with sporadic cancer; this phenomenon is involved in a stronger tendency to develop cancer.14 Recent studies have identified a panel of genes associated with greater vulnerability to developing FNMTC, specifically FOXE1, TPCSC2, MYH9, SRGAP1, HABP2, BRACA1, CHEK2, ATM, RASAL1, SRRM2, XRCC1, TIRT-1/NKX2.1 and PTCSC3.15 However, findings to date have not yet determined the impact of these genes on FNMTC development and prognosis, so genetic testing of families is not currently indicated.
Many studies comparing patients with FNMTC to patients with sporadic carcinoma (SC) have adopted a position in favour of a more aggressive approach in the familial group.16,17 Nevertheless, the form of presentation and prognosis thereof remain matters of debate, given those studies’ mixed nature and retrospective design.18 The primary objective of our study was to compare non-syndromic FNMTC and SC in terms of clinical and pathology-related characteristics. Differences between the subgroup of families with two or three members with DTC and the subgroup of patients with SC were also analysed, as were differences between first-generation and second-generation family members. Our study also sought to assess differences in initial treatment and determine the prognosis for FNMTC compared to SC.
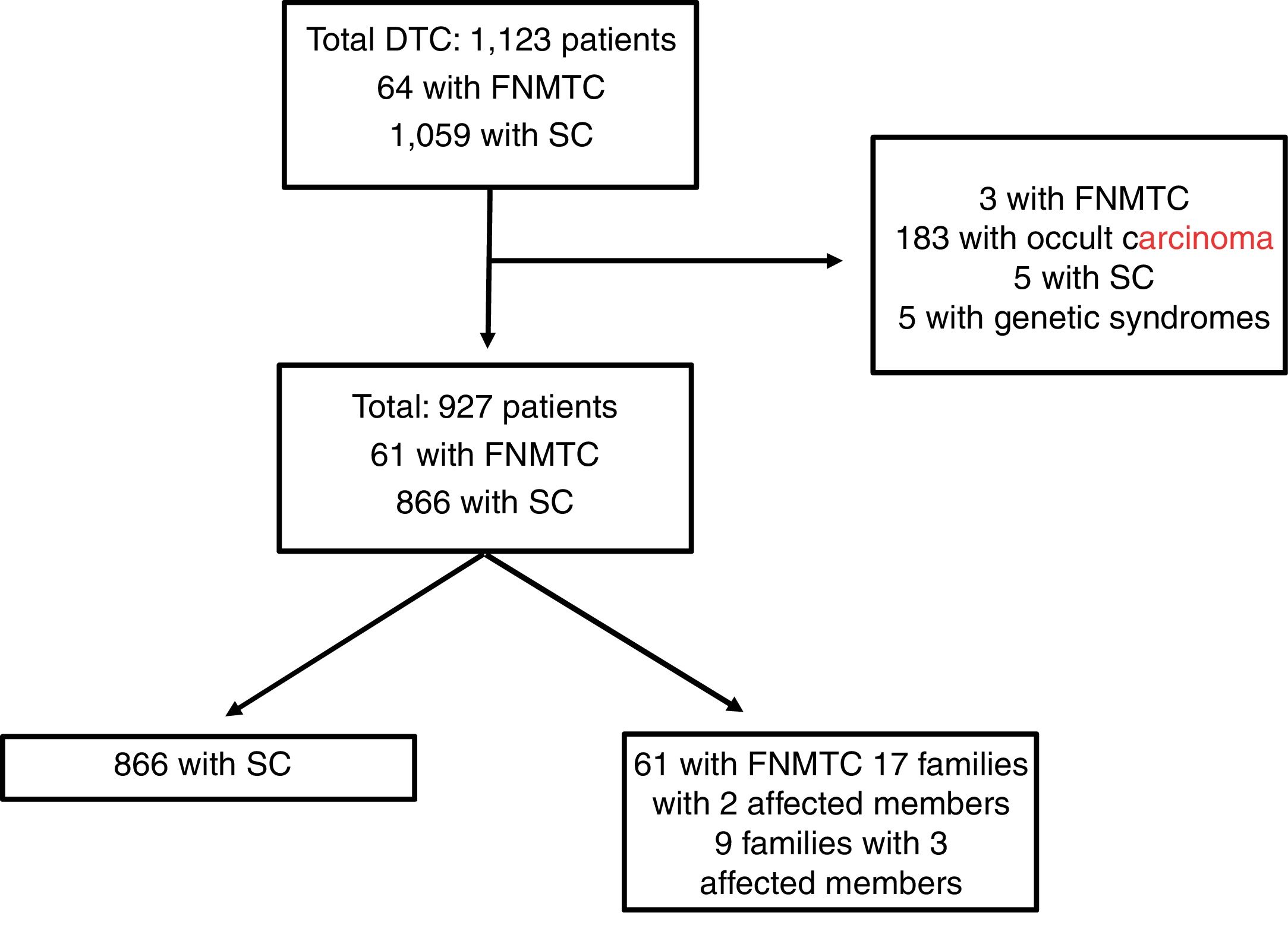
Materials and methodsPatients and methodsFig. 1 features a diagram with the patients included and excluded in the analysis. A total of 1123 patients included in the Navarre TC hospital cohort, diagnosed and treated at our centre between 1990 and 2018, with subsequent follow-up at our endocrinology clinic, were retrospectively evaluated. This study excluded patients with DTC as an incidental finding in surgery (n = 183), patients with genetic syndromes associated with TC (n = 5), five cases of SC due to missing data and three cases classified as FNMTC with the patients' relatives not in follow-up at our centre. Ultimately, the population in which the analysis was conducted consisted of 927 patients: 866 with SC and 61 with FNMTC.
 carcinoma.")
Patients with FNMTC were defined as patients with two or more first-degree relatives with a pathology diagnosis of TC of follicular origin. The first-degree relatives included were parents, siblings and offspring in follow-up at our centre. The 61 patients classified as FNMTC belonged to 26 different family groups; 17 families had two affected members (34 patients) and the remaining nine had three (27 patients).
Study protocolThe following were collected: demographic variables (age and gender), laboratory variables (antithyroglobulin antibody titre, baseline thyroglobulin, stimulated thyroglobulin, thyroid-stimulating hormone [TSH], free thyroxine [T4]), histopathological variables (size, lymph nodes, tumour pathology, multifocality, bilaterality, vascular invasion, lymph node size and extrathyroidal extension), treatment type, iodine-131 (I-131) dose and number of I-131 treatments. Data corresponding to post–I-131 body scan uptake, calculated risk of recurrence, dynamic stratification (excellent response, indeterminate response, biochemical incomplete response or structural incomplete response), disease-free survival and mortality due to thyroid disease were also analysed. The primary endpoint of the study was the presence of persistence or recurrence of disease at the end of the study or at the time of the patient's death. The histological characteristics of the tumour were determined by pathologists with experience in thyroid disease.
All patients were followed up at our centre at least once yearly. Follow-up was done by means of medical history, physical examination, determination of thyroglobulin and antithyroglobulin antibodies, and periodic neck ultrasound. Depending on clinical suspicion, stimulated thyroglobulin was determined with a body scan to detect persistence/recurrence and other imaging tests (X-ray, computed tomography, magnetic resonance imaging or positron emission tomography) were ordered.
A patient was deemed “disease-free” with a stimulated thyroglobulin level under 1 ng/mL, a negative result for antithyroglobulin antibodies, a normal body scan and a neck ultrasound with no pathological findings. The persistence and recurrence categories were unified and defined as evidence of disease one year after initial treatment (total thyroidectomy plus radioactive iodine), through detectable levels of thyroglobulin or a positive imaging test confirmed by means of cytology or surgery. Tumour staging was done at the time of diagnosis according to the seventh edition of the American Joint Committee on Cancer (AJCC) TNM classification.19
Statistical analysisContinuous variables were expressed in terms of mean ± standard deviation, and Student’s t test was used to compare these variables. Categorical variables were expressed in terms of numbers and percentages, and were compared using the chi-squared test. Comparison of disease-free survival between the different groups was done using Kaplan–Meier survival curves and the log-rank test. A p value under 0.05 was considered statistically significant. Statistical analysis was performed with the Stata version 12 software programme (Stata Statistical Software: College Station, TX: StataCorp LP).
ResultsBaseline characteristics appear in Table 1. A total of 927 patients, 61 of whom had FNMTC (6.6%), were included. The remaining 866 (93.4%) had SC. Mean follow-up was 9.7 ± 6.5 years. Mean patient age was 46.3 ± 15.0 years, with a higher prevalence in women (78%).
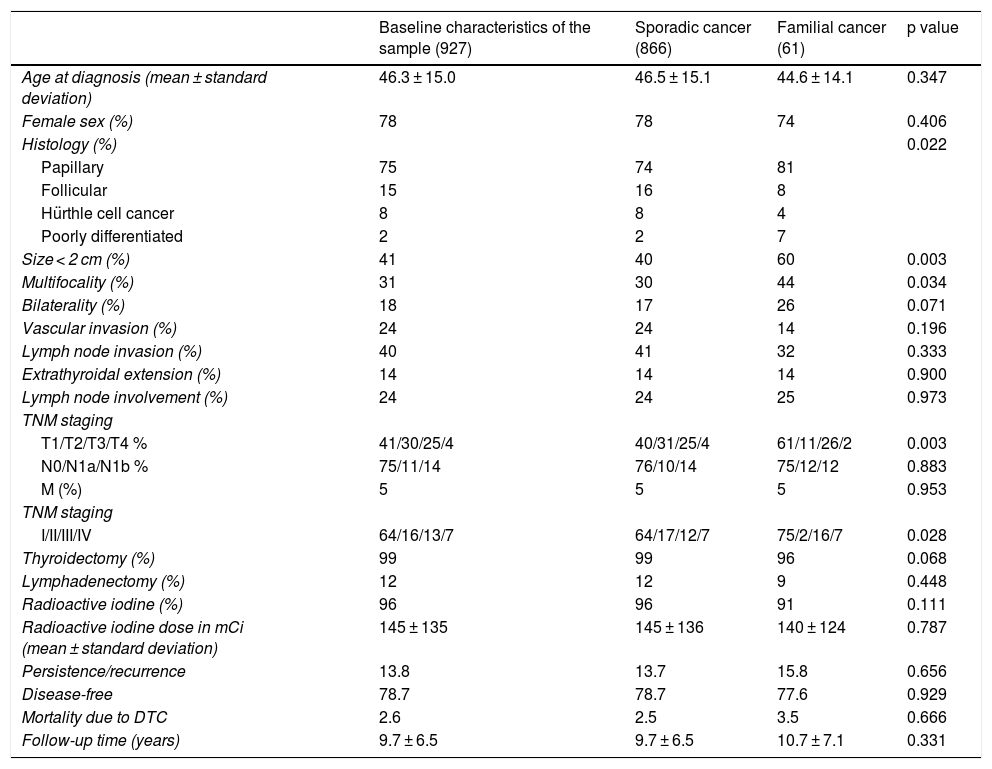
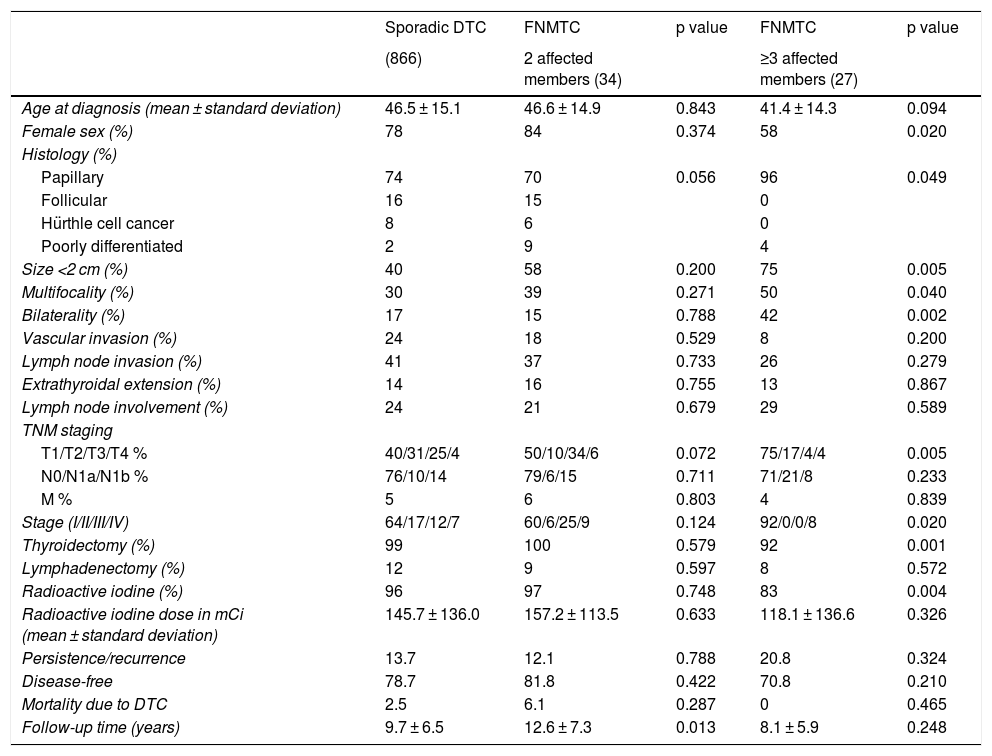
Description of the sample and subgroups. Comparison of clinical and pathology-related characteristics of sporadic DTC versus familial non-medullary DTC.
| Baseline characteristics of the sample (927) | Sporadic cancer (866) | Familial cancer (61) | p value | |
|---|---|---|---|---|
| Age at diagnosis (mean ± standard deviation) | 46.3 ± 15.0 | 46.5 ± 15.1 | 44.6 ± 14.1 | 0.347 |
| Female sex (%) | 78 | 78 | 74 | 0.406 |
| Histology (%) | 0.022 | |||
| Papillary | 75 | 74 | 81 | |
| Follicular | 15 | 16 | 8 | |
| Hürthle cell cancer | 8 | 8 | 4 | |
| Poorly differentiated | 2 | 2 | 7 | |
| Size < 2 cm (%) | 41 | 40 | 60 | 0.003 |
| Multifocality (%) | 31 | 30 | 44 | 0.034 |
| Bilaterality (%) | 18 | 17 | 26 | 0.071 |
| Vascular invasion (%) | 24 | 24 | 14 | 0.196 |
| Lymph node invasion (%) | 40 | 41 | 32 | 0.333 |
| Extrathyroidal extension (%) | 14 | 14 | 14 | 0.900 |
| Lymph node involvement (%) | 24 | 24 | 25 | 0.973 |
| TNM staging | ||||
| T1/T2/T3/T4 % | 41/30/25/4 | 40/31/25/4 | 61/11/26/2 | 0.003 |
| N0/N1a/N1b % | 75/11/14 | 76/10/14 | 75/12/12 | 0.883 |
| M (%) | 5 | 5 | 5 | 0.953 |
| TNM staging | ||||
| I/II/III/IV | 64/16/13/7 | 64/17/12/7 | 75/2/16/7 | 0.028 |
| Thyroidectomy (%) | 99 | 99 | 96 | 0.068 |
| Lymphadenectomy (%) | 12 | 12 | 9 | 0.448 |
| Radioactive iodine (%) | 96 | 96 | 91 | 0.111 |
| Radioactive iodine dose in mCi (mean ± standard deviation) | 145 ± 135 | 145 ± 136 | 140 ± 124 | 0.787 |
| Persistence/recurrence | 13.8 | 13.7 | 15.8 | 0.656 |
| Disease-free | 78.7 | 78.7 | 77.6 | 0.929 |
| Mortality due to DTC | 2.6 | 2.5 | 3.5 | 0.666 |
| Follow-up time (years) | 9.7 ± 6.5 | 9.7 ± 6.5 | 10.7 ± 7.1 | 0.331 |
Table 1 compares the characteristics of the patients with FNMTC and with SC. No significant differences were detected in relation to age at diagnosis (mean 44.6 ± 14.1 years versus 46.5 ± 15.1 years; p = 0.347) or female sex (74% versus 78%; p = 0.406). A higher rate of multifocal presentation was seen in the FNMTC group (44% versus 30%; p = 0.034). The predominant histological variant in both groups was the papillary subtype, although this histological variant was represented at a higher rate in cases of FNMTC (81% versus 74%; p = 0.022). Tumour size was under 2 cm in 60% of patients with FNMTC versus 40% of patients with SC (p = 0.003), leading to significantly lower TNM staging (p = 0.003). We found no significant differences with respect to bilateral disease presentation (26% versus 17%; p = 0.071), lymph node invasion (32% versus 41%; p = 0.333), vascular invasion (14% versus 24%; p = 0.196) or extrathyroidal extension (14% versus 14%; p = 0.900).
No significant differences were found in terms of initial approach to treatment. Total thyroidectomy was performed in one or two operations in 96% of cases of FNMTC versus 99% of cases of SC (p = 0.068). Cervical lymphadenectomy was required in 9% and in 12% of patients, respectively (p = 0.448). I-131 was subsequently administered in 91% of patients with FNMTC and 96% of patients with SC (p = 0.111), with a comparable cumulative dose of 140 ± 124 millicuries (mCi) in FNMTC and 145 ± 136 mCi in SC (p = 0.787).
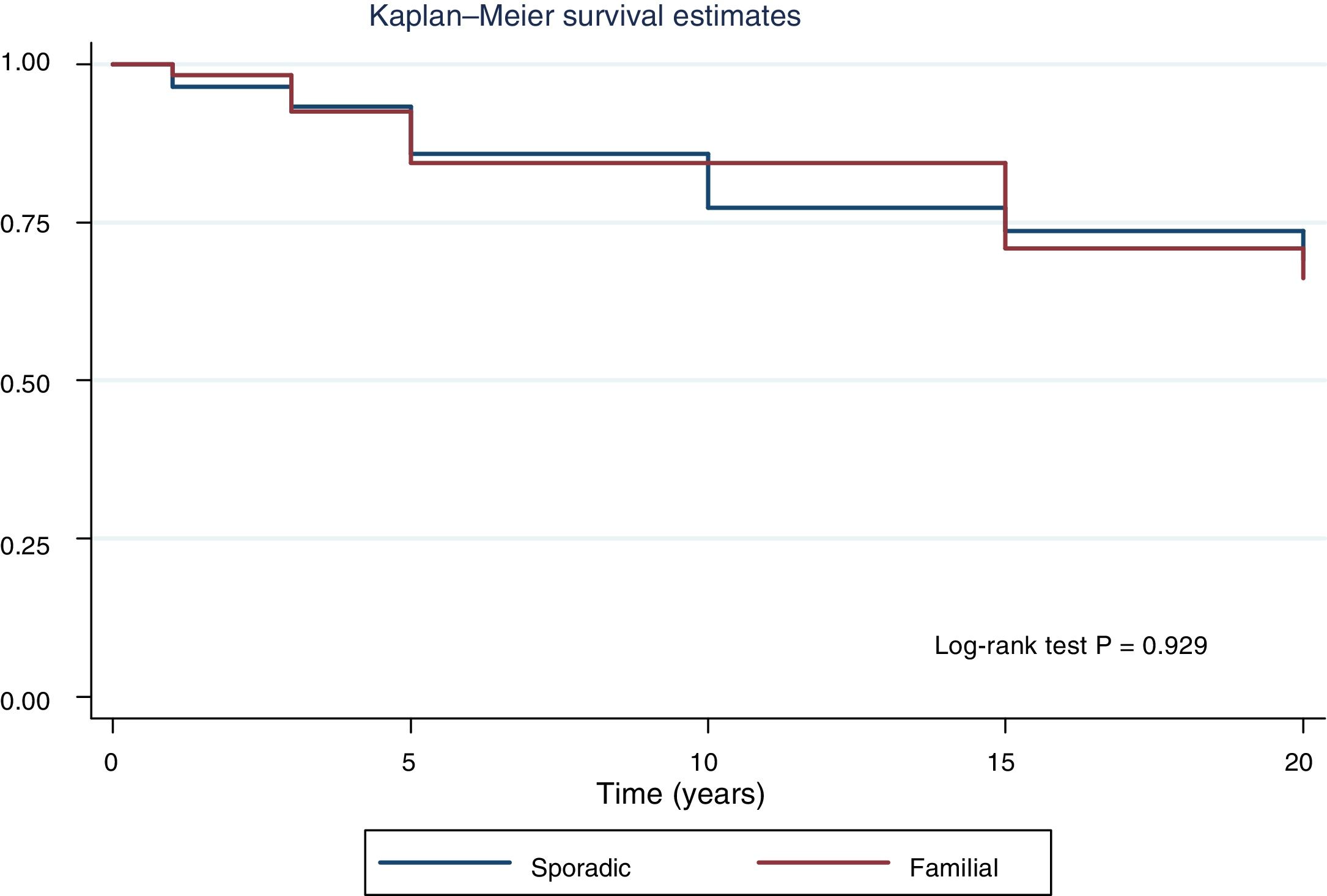
Fig. 2 shows the disease-free survival curves for both groups. Disease-free survival at the end of follow-up was similar (77.6% versus 78.7%; p = 0.929). In addition, no differences were found in rates of persistence/recurrence in FNMTC versus SC (15.8% versus 13.7%; p = 0.656) or in mortality resulting from carcinoma, which was around 3.5% in FNMTC versus 2.5% in SC (p = 0.331).

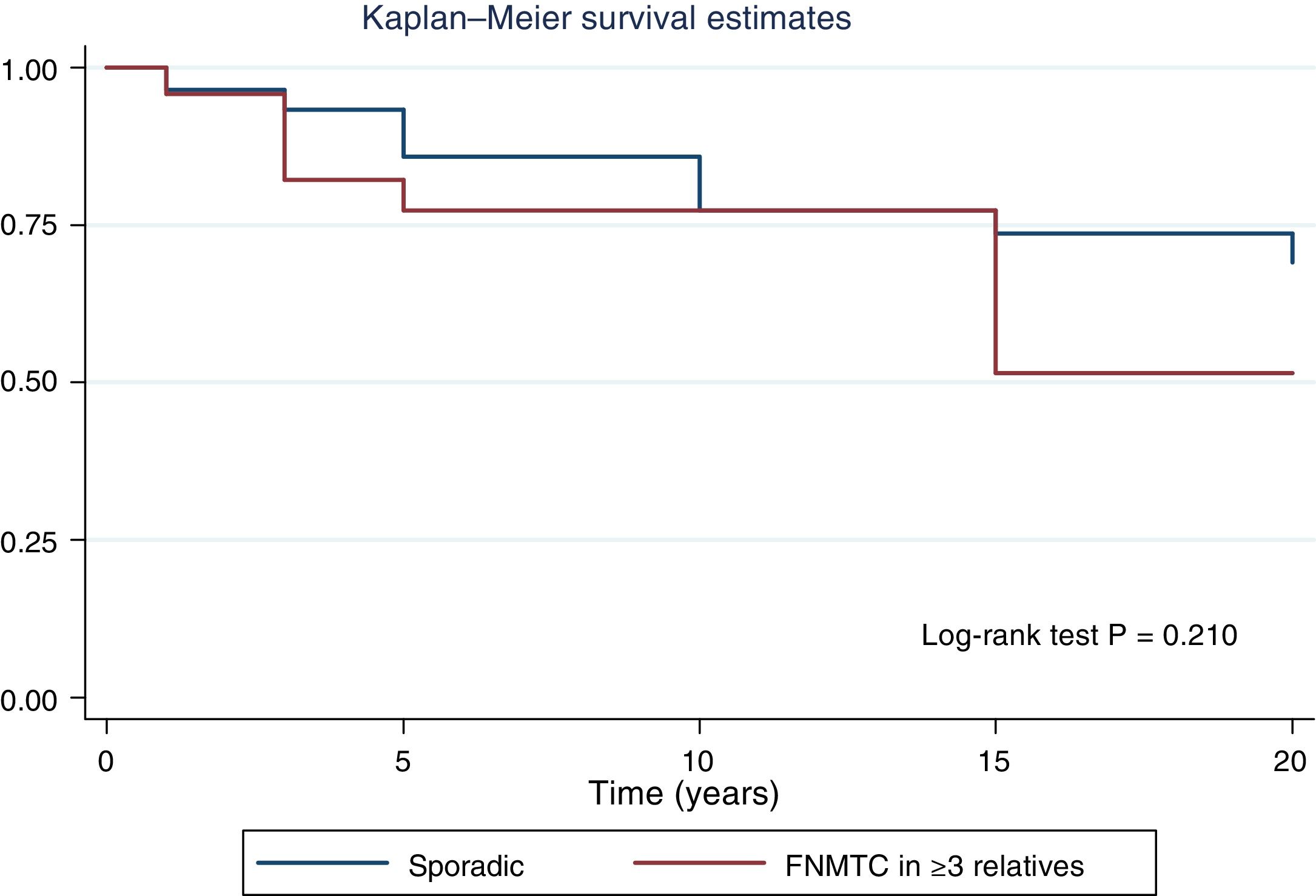
Table 2 details the subanalysis with the differences found when cases of FNMTC in families with two or three affected members were compared to cases of SC. It was found that, in the group with three or more affected relatives, tumours were smaller in size (15.6 ± 14.3 mm versus 26.5 ± 16.9 mm; p = 0.002), with greater multifocality (50% versus 30%; p = 0.040), bilaterality (42% versus 17%; p = 0.002) and a higher rate of males (42% versus 22%; p = 0,020), and that the most common subtype was the papillary subtype (96% versus 74%; p = 0.049). Furthermore, in FNMTC with three affected family members, there were lower numbers of thyroidectomies (92% versus 99%; p = 0.001) and treatments with radioactive iodine (83% versus 96%; p = 0.004). With respect to prognosis, incidence of persistence/recurrence, disease-free survival (Fig. 3) and mortality resulting from carcinoma were comparable in the two groups.
Comparison of clinical and pathology-related characteristics of sporadic DTC versus FNMTC in families with 2 affected members and versus FNMTC in families with ≥3 affected members.
| Sporadic DTC | FNMTC | p value | FNMTC | p value | |
|---|---|---|---|---|---|
| (866) | 2 affected members (34) | ≥3 affected members (27) | |||
| Age at diagnosis (mean ± standard deviation) | 46.5 ± 15.1 | 46.6 ± 14.9 | 0.843 | 41.4 ± 14.3 | 0.094 |
| Female sex (%) | 78 | 84 | 0.374 | 58 | 0.020 |
| Histology (%) | |||||
| Papillary | 74 | 70 | 0.056 | 96 | 0.049 |
| Follicular | 16 | 15 | 0 | ||
| Hürthle cell cancer | 8 | 6 | 0 | ||
| Poorly differentiated | 2 | 9 | 4 | ||
| Size <2 cm (%) | 40 | 58 | 0.200 | 75 | 0.005 |
| Multifocality (%) | 30 | 39 | 0.271 | 50 | 0.040 |
| Bilaterality (%) | 17 | 15 | 0.788 | 42 | 0.002 |
| Vascular invasion (%) | 24 | 18 | 0.529 | 8 | 0.200 |
| Lymph node invasion (%) | 41 | 37 | 0.733 | 26 | 0.279 |
| Extrathyroidal extension (%) | 14 | 16 | 0.755 | 13 | 0.867 |
| Lymph node involvement (%) | 24 | 21 | 0.679 | 29 | 0.589 |
| TNM staging | |||||
| T1/T2/T3/T4 % | 40/31/25/4 | 50/10/34/6 | 0.072 | 75/17/4/4 | 0.005 |
| N0/N1a/N1b % | 76/10/14 | 79/6/15 | 0.711 | 71/21/8 | 0.233 |
| M % | 5 | 6 | 0.803 | 4 | 0.839 |
| Stage (I/II/III/IV) | 64/17/12/7 | 60/6/25/9 | 0.124 | 92/0/0/8 | 0.020 |
| Thyroidectomy (%) | 99 | 100 | 0.579 | 92 | 0.001 |
| Lymphadenectomy (%) | 12 | 9 | 0.597 | 8 | 0.572 |
| Radioactive iodine (%) | 96 | 97 | 0.748 | 83 | 0.004 |
| Radioactive iodine dose in mCi (mean ± standard deviation) | 145.7 ± 136.0 | 157.2 ± 113.5 | 0.633 | 118.1 ± 136.6 | 0.326 |
| Persistence/recurrence | 13.7 | 12.1 | 0.788 | 20.8 | 0.324 |
| Disease-free | 78.7 | 81.8 | 0.422 | 70.8 | 0.210 |
| Mortality due to DTC | 2.5 | 6.1 | 0.287 | 0 | 0.465 |
| Follow-up time (years) | 9.7 ± 6.5 | 12.6 ± 7.3 | 0.013 | 8.1 ± 5.9 | 0.248 |
Data in bold are statistically significant (p < 0.05).

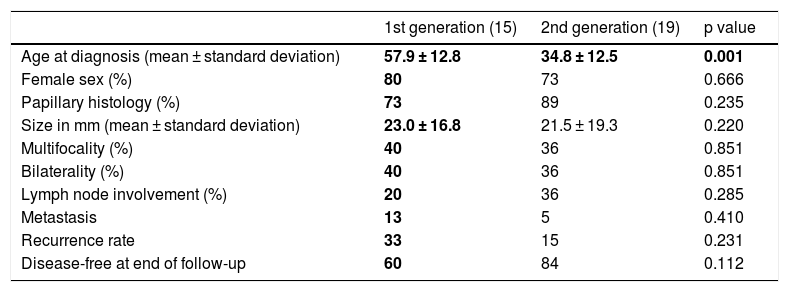
Of the 61 cases of FNMTC, 34 (56%) had a parent–child relationship and 27 had a sibling relationship. Analysis of the 34 cases of parents and children revealed that 15 patients belonged to the first generation and that 19 were children and therefore belonged to the second generation. When the two generations were compared in Table 3, age at diagnosis was seen to be 23.1 years younger in second-generation members compared to first-generation members (34.8 ± 12.5 years versus 57.9 ± 12.8 years; p = 0.001). Despite this, size, pathology-related characteristics and prognosis were similar (p > 0.05).
Comparison of clinical, pathology-related and prognostic characteristics between the first and the second generation.
| 1st generation (15) | 2nd generation (19) | p value | |
|---|---|---|---|
| Age at diagnosis (mean ± standard deviation) | 57.9 ± 12.8 | 34.8 ± 12.5 | 0.001 |
| Female sex (%) | 80 | 73 | 0.666 |
| Papillary histology (%) | 73 | 89 | 0.235 |
| Size in mm (mean ± standard deviation) | 23.0 ± 16.8 | 21.5 ± 19.3 | 0.220 |
| Multifocality (%) | 40 | 36 | 0.851 |
| Bilaterality (%) | 40 | 36 | 0.851 |
| Lymph node involvement (%) | 20 | 36 | 0.285 |
| Metastasis | 13 | 5 | 0.410 |
| Recurrence rate | 33 | 15 | 0.231 |
| Disease-free at end of follow-up | 60 | 84 | 0.112 |
Data in bold are statistically significant (p < 0.05).
FNMTC has been defined as a differentiated entity accounting for 5%–10% of all cases of DTC. This study found the prevalence of FNMTC to be 6.6% (61/927), at the lower limit of the rates reported in other series8,13,20 and far from the 9.6% in other registries, such as registries from South Korea and North America.20,21 A lack of clinical and genetic markers in DTC renders FNMTC indistinguishable from SC. Given DTC's high incidence and survival rates, an estimated 31%–38% of patients with carcinoma who have two affected relatives and 94% of patients with carcinoma who have three affected relatives actually have FNMTC and not SC.8 This in our sample could mean that 34 of the 61 patients are true cases with familial aggregation.
Some studies have reported more aggressive behaviour on the part of FNMTC and a worse prognosis compared to SC, whereas others have found the opposite. The characteristic most commonly reported across different studies has been greater multifocality and bilaterality, consistent with our cohort. Zhang et al.7 found rates of multifocality in FNMTC compared to SC of 60% versus 40% and bilaterality of 44% versus 29%, respectively (p < 0.05). A case–control study by Cao et al.17 detected rates of multifocality and bilaterality in the family group of 54% and 44%, respectively, compared to 39% and 28% in cases of SC (p < 0.05).
The smaller tumour size reported in our study is more subject to debate according to the literature consulted. In our case, it even meant lower TNM staging and less aggressive treatments in the group with three affected family members. Lakis et al.,16 in a study of 78 cases of FNMTC, found a higher proportion of T1 tumours. However, a 2015 meta-analysis that included 12 studies with patients from Asia, North America and Europe22 found no differences with respect to size. In our cohort, overall, the proportion of tumours under 2 cm was 41% — well below the 75% reported by Lakis et al.16 The larger tumour size in our series might have been due to its exclusion of cases of microcarcinoma detected incidentally in benign thyroid surgery. Therefore, cases of occult DTC measuring less than centimetre and discovered unexpectedly in examination of the surgical specimen by pathology were not included.23 This heightened the differences between the family group and the sporadic group, with FNMTC having a higher rate of cases of T1 carcinoma (61%) in comparison to SC (40%) (p = 0.003).
Zhang et al.24 found cases of FNMTC to have higher rates of lymph node disease (52.6% versus 33.3%; p < 0.05) and extrathyroidal extension (64.1% versus 48.5%; p < 0.05). In our sample, no significant differences were found along these two dimensions.
A growing body of studies has found FNMTC to be more aggressive, especially in the group with three affected family members. Alsanea et al.25 detected higher rates of recurrence in FNMTC (44% versus 17%; p < 0.05) along with lower rates of disease-free survival compared to SC. McDonald et al.26 determined that groups with three or more relatives with DTC underwent more repeat operations (p = 0.05) or required additional doses of radioactive iodine (p = 0.03) and had more remote metastases (p = 0.003) and deaths (p = 0.01).
According to our data, despite greater multifocality (p = 0.040) and bilaterality (p = 0.002), which could be an early marker of FNMTC aggressiveness, prognosis in the form of persistence/recurrence (p = 0.324) and mortality due to DTC (p = 0.465) was comparable, with no evidence of a worse clinical course. No screening that might have accounted for such results was performed, but heightened awareness of DTC in the family group could have been associated with diagnosis in an earlier stage and therefore less aggressive treatment with a comparable prognosis.
Most studies have found a younger age of presentation of FNMTC compared to SC.7 Moses and Weng20 reported age at diagnosis to be five years younger on average. However, this figure could be affected by the phenomenon of anticipation of the second generation published by Capezzone et al.2 in 2008. They demonstrated in an Italian cohort that DTC diagnosis in the second generation is anticipated in comparison to that generation's parents. This was subsequently confirmed by a study by Zhou et al.,27 among others. In our sample, the age of presentation of FNMTC was 23.1 years younger in the second generation. If this anticipation were merely a reflection of an early diagnosis, other differences such as a smaller tumour size or lesser aggressiveness could be expected. Yet, all other variables were comparable, supporting the notion of the existence of this genetic phenomenon seen in other conditions, both benign and malignant.
We found a higher rate of males in the group with three affected family members. This finding was also reported by Lakis et al.16 with a cohort of 78 cases of FNMTC, as well as Cao et al.17 in a case–control study with 372 cases. This was consistent with the autosomal dominant inheritance pattern suspected in this disease. Given this genetic factor, the predominance of females in SC disappears in FNMTC, with a larger number of males. DTC, specifically its papillary subtype, shows a higher risk when a first-degree relative or twin is diagnosed, as it is among the cancers possessed of the highest standardised incidence ratios (SIRs), with values of 3.21 and 6.24, respectively.28 It is likely due to a group of genes with an autosomal dominant inheritance pattern and incomplete penetrance that is more likely to be passed down by mothers (SIR 4.32).29 The higher overall incidence of papillary DTC in women suggests that oestrogenic stimulation of the thyroid gland plays a role in promoting progression or malignant transformation.30 At the onset of puberty, the incidence of DTC increases only in the female gender and drops again after menopause, possibly due to a membrane-bound oestrogen receptor-mediated effect of growth promotion.31
As regards the mechanism of transmission, Tavarelli et al.,32 in a retrospective study with 151 individuals from 74 families, found it to most commonly occur between siblings (62.2%), followed by inheritance from the mother (26.5%) and finally inheritance from the father (7.3%). In our sample, 27 patients (44%) had a sibling relationship and 34 (56%) patients had a parent–child relationship (three of them had a father–child relationship and 12 had a mother–child relationship).
Our results were consistent with prior studies having concluded that differences between SC and FNMTC could be attributed to the group of relatives with three affected family members. This indicates that families with three or more members with FNMTC constitute true familial cases and that patients with two family members may represent a grouping of two sporadic cases.
The main strength of the study was that it compiled all patients diagnosed with DTC in a geographic area over a relatively long period of time, reflecting the population with which we work on a day-to-day basis. Diagnostic screening was not performed and treatment was comparable in the two groups, boosting the validity of the results in relation to prognosis.
The study also had several limitations. First, given the low prevalence of this disease, the sample size of the FNMTC cohort was modest. The data came from a single Autonomous Community of Spain and were analysed retrospectively, meaning that generalisation of its conclusions may carry bias. Finally, given the lack of markers and genes specifically confirming a case of FNMTC, some percentage of cases with two members diagnosed with DTC classified as familial are, in reality, two cases of SC that happen to occur in a single family (due to the relatively high incidence of DTC). However, this adds to the value of the differences found, given that including these cases could only dilute the aggressiveness demonstrated.
In conclusion, FNMTC represents a differentiated entity within DTC. Families with three or more members with FNMTC constitute true familial cases with smaller tumours, higher rates of multifocality and bilaterality and higher rates of representation of the papillary subtype, and occurring more often in men. However, this study found no differences with respect to prognosis or lymph node involvement; therefore, the data are insufficient to justify more aggressive treatment. This means that the initial treatment proposed should be similar to that in SC, depending on risk and baseline TNM staging. Regarding the surgical approach, the higher rate of bilaterality should be taken into account. Patients with FNMTC do not require more intensive follow-up, since recurrence and persistence rates are similar, and their life expectancy is not determined by the cancer itself. Despite the aggregation and the up to three times higher risk of DTC in individuals with more than three affected family members, at present, there is no indication to engage in screening for early detection, given its good prognosis. Perhaps, in line with the data presented, it would be reasonable to study relatives of the subgroup with three or more members diagnosed with DTC. In these high-risk families, the phenomenon of anticipation in the second generation should be taken into account to determine the age at which testing is to be performed.
Conflicts of interestThe authors declare no conflicts of interest.
The authors would like to thank all the members of the multidisciplinary thyroid cancer committee at Complejo Hospitalario de Navarra [Navarre Hospital Complex], made up of the departments of Endocrinology and Nutrition, General Surgery, Pathology, Diagnostic Imaging and Medical Oncology.
Please cite this article as: de Carlos Artajo J, Irigaray Echarri A, García Torres J, Pineda Arribas JJ, Ernaga Lorea A, Eguílaz Esparza N, et al. Características clínicas y pronósticas del carcinoma familiar de tiroides no medular. Endocrinol Diabetes Nutr. 2022;69:262–270.
