




Las dislipemias familiares son trastornos del metabolismo de los lípidos que tienden a agruparse en determinados núcleos familiares. En ocasiones son de causa genética, pero en gran parte de los casos son multifactoriales, y el ambiente compartido juega un papel importante. Es el caso de la dislipemia familiar más común, la hiperlipemia combinada multifactorial, antes llamada hiperlipemia familiar combinada, donde el componente ambiental, especialmente el sobrepeso y la dieta, influyen en el fenotipo final de los pacientes.
Estas dislipemias siguen distintos patrones de presentación en la familia y pueden estar originadas por enfermedades monogénicas, poligénicas o ser de origen multifactorial. En general, se prefiere reservar el término familiar a las dislipemias producidas por defectos en un único gen, como ocurre en la hipercolesterolemia familiar (HF).
El fenotipo lipídico puede presentar un alto grado de solapamiento entre los diferentes tipos de dislipemia, condicionados por la gravedad del defecto genético, la presencia de otras variantes genéticas asociadas y la interacción con el ambiente. El diagnóstico clínico puede ser complejo, ya que existen pocos datos fenotípicos patognomónicos, entre los que encontramos los xantomas tendinosos o el arco corneal en la HF, los xantomas palmares, planos o tuberosos en la disbetalipoproteinemia o las pancreatitis de repetición desde la infancia, en la quilomicronemia familiar por déficit de lipoproteína lipasa. Sin embargo, la ausencia de estos estigmas no descarta la enfermedad. Por ello, el estudio genético resulta de vital importancia para lograr una aproximación lo más exacta posible al diagnóstico de estas enfermedades.
La dislipemia monogénica más frecuente es la HF, generalmente en heterocigosis (HF heterocigota o HFHe) y está producida mutaciones en el gen que codifica el receptor de LDL (LDLR) (85-90% de los casos), la apolipoproteína B (APOB) (5%), mutaciones con ganancia de función (GOF) en el gen de la proproteína convertasa subtilisina-kexina tipo 9 (PCSK9) (1-3%), de la apolipoproteínaE (APOE) (variante p.Leu167del) o del de la proteína adaptadora tipo 1 del receptor de LDL (LDLRAP1), este último de herencia autosómica recesiva1. La HFHe se caracteriza por niveles muy elevados de colesterol unido a lipoproteínas de baja densidad (c-LDL), generalmente por encima de 250mg/dl, trasmisión vertical de la hipercolesterolemia con aproximadamente el 50% de los familiares de primer grado afectos, enfermedad cardiovascular (ECV) prematura, arco corneal antes de los 45años y en algunos casos xantomas tendinosos, principalmente en el tendón de Aquiles1,2.
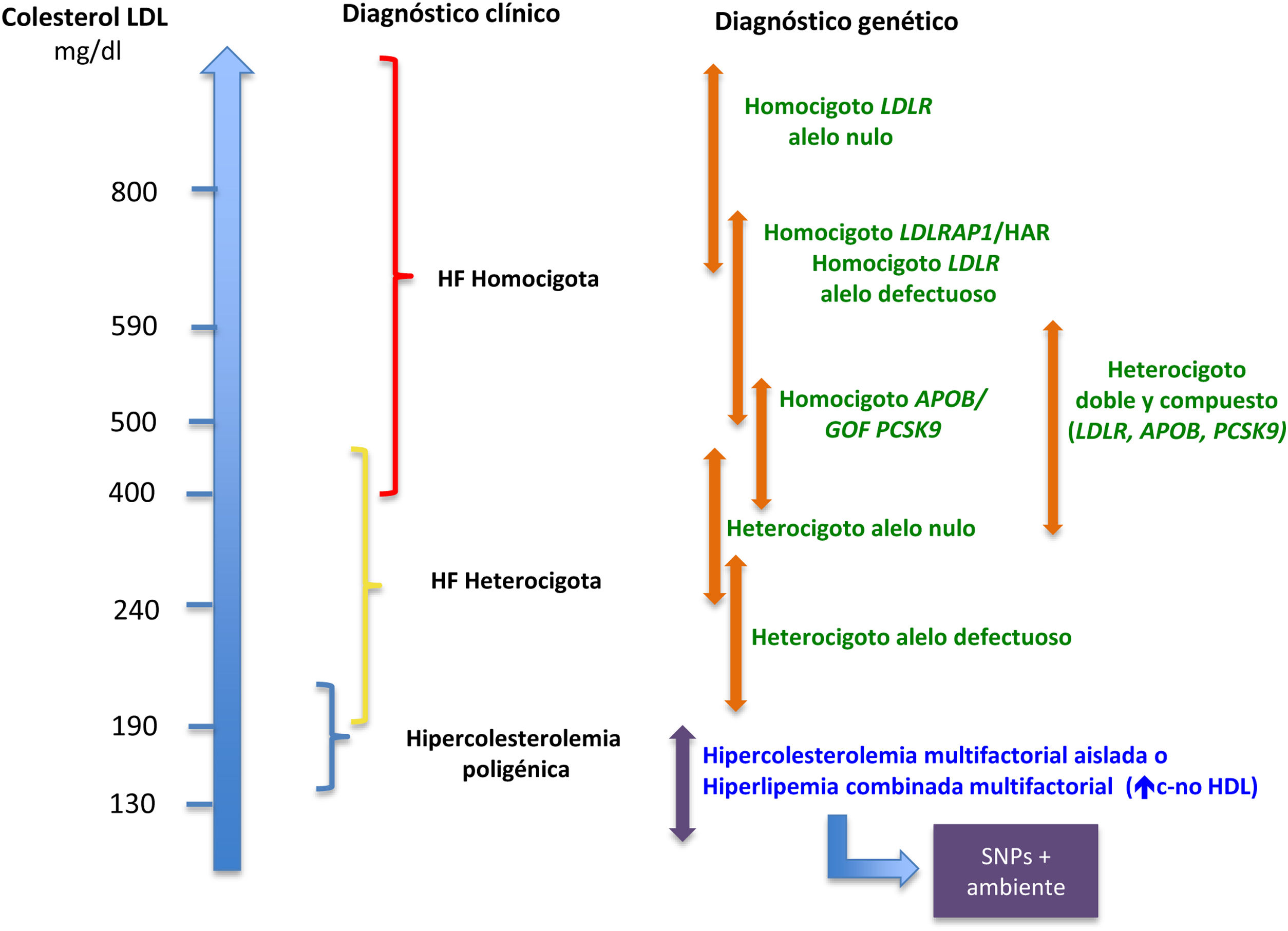
En el gen LDLR se han descrito más de 4.000 variantes alélicas3, que afectan de forma distinta a la función del receptor de LDL (LDLr), y esto se traduce en una variabilidad fenotípica importante4 (fig. 1). Existen mutaciones más graves, en las que no se produce LDLr (alelos nulos o de clase1), asociadas a niveles más altos de c-LDL, peor respuesta al tratamiento y más ECV, y mutaciones más leves (alelo defectuoso o no de clase1), en las que hay proteína funcional, pero está alterada.

Relación genotipo-fenotipo en la hipercolesterolemia.
APOB: gen de la apolipoproteína B; c: colesterol; GOF: ganancia de función; HAR: hipercolesterolemia autosómica recesiva; HDL: lipoproteínas de alta densidad; HF: hipercolesterolemia familiar; LDL: lipoproteínas de baja densidad; LDLR: gen del receptor de LDL; LDLRAP1: gen de la proteína adaptadora tipo 1 del receptor de LDL; PCSK9: gen de la proproteína convertasa subtilixina-kexina tipo 9; SNP: single nucleotide polymorphism.
Por otro lado, también el gen implicado tiene consecuencias sobre el fenotipo. La gravedad del mismo suele seguir el orden LDLR >APOB >PCSK9. Y dentro de las mutaciones GOF de PCSK9 también hay variabilidad, según se presente la variante p.(Asp374Tyr), que genera un fenotipo más grave, u otras5.
Aunque existe una amplia correlación genotipo-fenotipo, y a mayor gravedad del defecto genético peor fenotipo clínico, hay sujetos con niveles no tan elevados o incluso normales de c-LDL a pesar de la presencia de mutación. De igual forma, existen sujetos con fenotipo clínico de HF con diagnóstico genético negativo. Esto es, en parte, por la sensibilidad y la especificidad limitadas de las escalas clínicas de diagnóstico, como los criterios de las Redes Clínicas de Lípidos Holandesas2, en las que nos encontramos entre un 20 y un 40% de los casos con diagnóstico clínico de HF y estudio genético negativo1. En general, a mayores niveles de c-LDL habrá más probabilidad de encontrar una mutación en un gen causal de HF6, por lo que la recomendación para realizar el estudio genético ante la sospecha de una forma monogénica serían niveles de c-LDL >240mg/dl7.
Dentro del espectro de la HF también existen formas homocigotas o HF bialélica, menos frecuentes, que comprenden dos mutaciones en los genes mencionados8. Estas formas son más graves y presentan niveles de c-LDL por encima de 400mg/dl, ECV más prematura, xantomas precoces, incluso desde la infancia, y suelen estar afectos ambos progenitores de HFHe9. En la HFHe existe una correlación genotipo-fenotipo marcada, y si sospechamos esta entidad el diagnóstico genético precoz es prioritario, porque son sujetos con muy mal pronóstico cardiovascular si no se instaura un tratamiento temprano.
Otra forma de hipercolesterolemia primaria es la hipercolesterolemia poligénica (HP), que comprende la suma de distintas mutaciones puntuales o polimorfismos (single nucleotide polymorphism [SNP]) en genes implicados en el metabolismo lipídico, que de forma aislada tienen un efecto pequeño sobre las concentraciones de c-LDL, pero su conjunto genera hipercolesterolemias primarias con valores de c-LDL similares a veces a los de formas monogénicas. Estos SNP se agrupan en escalas denominadas puntuaciones de riesgo poligénico (PRS)10 y normalmente se consideran HP por encima del percentil 75 o 90 de la distribución de esa PRS. Las PRS suponen un avance muy importante en la caracterización genética de las hipercolesterolemias primarias. La mayor parte de los casos de fenotipos de HF con estudio genético negativo son formas poligénicas graves. Incluso, en ocasiones, el fenotipo más grave observado en la HF se debe a la presencia de variantes poligénicas o comunes, que, sumadas a la mutación monogénica, generan un empeoramiento de fenotipo lipídico. De hecho, estos pacientes con mutación monogénica y PRS elevado tienen peor pronóstico cardiovascular11.
Por último, tenemos la hipercolesterolemia multifactorial aislada o hiperlipemia combinada multifactorial. En ella suelen encontrarse niveles de c-LDL ≥130mg/dl o colesterol no-HDL ≥160mg/dl como resultado de la interacción de factores poligénicos poco conocidos y ambientales, como el sobrepeso y la obesidad, que actúan como desencadenantes del fenotipo lipídico. Generalmente, esta suma de ambiente y genética suele estar presente en la misma familia, por lo que el fenotipo puede aparecer en distintas generaciones, simulando una forma monogénica, pero con fenotipos variables: a veces se presenta como hipercolesterolemia aislada y otras veces como una hiperlipemia combinada. Esta entidad ha sido ampliamente estudiada y se ha descartado su origen monogénico1.
Como conclusión, podemos decir que las dislipemias familiares tienen una base genética monogénica, poligénica o multifactorial, y que el tipo de defecto genético marcará la gravedad del fenotipo en la mayoría de las ocasiones. Sin embargo, muchas veces la suma de variantes comunes en genes implicados en el metabolismo de los lípidos puede explicar en gran medida la variabilidad en la expresión fenotípica de la HF. Por todo esto, es de vital importancia la realización del estudio genético, incluyendo, si fuera posible, la realización de PRS para evaluar el componente poligénico. Esto nos va a permitir definir de forma más precisa la dislipemia, valorar su pronóstico, realizar el estudio a familiares y poder implementar el tratamiento adecuado.
FinanciaciónEste trabajo está financiado por un Proyecto de Investigación en Salud (PI20/00846) y un contrato de intensificación de la actividad investigadora (INT21/00032) del Instituto de Salud Carlos III (ISCIII).
Conflicto de interesesLos autores declaran no tener conflicto de intereses.
