




The association between diabetes and cancer was hypothesized almost one century ago. Today, a vast number of epidemiological studies support that obese and diabetic populations are more likely to experience tissue-specific cancers, but the underlying molecular mechanisms remain unknown. Obesity, diabetes, and cancer share many hormonal, immune, and metabolic changes that may account for the relationship between diabetes and cancer. In addition, antidiabetic treatments may have an impact on the occurrence and course of some cancers. Moreover, some anticancer treatments may induce diabetes. These observations aroused a great controversy because of the ethical implications and the associated commercial interests. We report an epidemiological update from a mechanistic perspective that suggests the existence of many common and differential individual mechanisms linking obesity and type 1 and 2 diabetes mellitus to certain cancers. The challenge today is to identify the molecular links responsible for this association. Classification of cancers by their molecular signatures may facilitate future mechanistic and epidemiological studies.
Hace casi un siglo que se hipotetizó la asociación entre la diabetes y el cáncer. Hoy, numerosos estudios epidemiológicos sostienen que las poblaciones con obesidad y/o diabetes poseen una mayor predisposición a padecer cáncer en órganos específicos. Los mecanismos moleculares subyacentes se desconocen. Las alteraciones metabólicas, hormonales e inmunológicas que comparten la obesidad, la diabetes y el cáncer pueden contribuir a justificar la relación existente. Por otra parte, la influencia de los tratamientos antidiabéticos en la aparición/evolución de algunos cánceres y la inducción de la diabetes por los tratamientos antineoplásicos han despertado una gran controversia debido a las implicaciones éticas y los intereses comerciales asociados. Esta actualización de los datos epidemiológicos presenta un enfoque mecanístico que sugiere la existencia de múltiples mecanismos comunes y diferenciales que asocian la obesidad y la diabetes tipo 1 y tipo 2 a ciertos cánceres. Identificar los mecanismos responsables de la asociación diabetes-cáncer es un reto de la investigación actual; la clasificación de los cáncer por sus firmas moleculares podría facilitar futuros estudios mecanísticos y epidemiológicos.
Obesity, diabetes, and cancer are metabolic disorders. Obesity predisposes to diabetes, and both obesity and diabetes are risk factors for many types of cancer.1 According to the World Health Organization (WHO), 39% of the world's adult population are overweight, and more than 13% are obese; these figures have doubled since 1980. Among adults, 9% are diabetic; this figure is expected to double by 2030 (http://apps.who.int/iris/bitstream/10665/148114/1/9789241564854_eng.pdf). Similarly, the WHO expects a 70% increase in cases of cancer by 2030 (http://publications.iarc.fr/Non-Series-Publications/World-Cancer-Reports/World-Cancer-Report-2014).
Both type 1 (T1DM) and type 2 diabetes mellitus (T2DM) share with cancer hormonal (defects in the insulin/IGF-1 and leptin/adiponectin axes),2 metabolic (affecting carbohydrates and lipids), and immune (increased circulating inflammatory cytokines)3 changes.
Human health epidemiological studies try to elucidate the impact of disease, social and environmental factors, etc. on the population's health. One way to measure this impact is to estimate the increase or decrease in the relative risk (RR) of developing another condition. This requires large populations of cases and controls (prospective or retrospective), but has the advantage that the results may be generalized to the overall population. The drawback is that in order for a study to achieve statistical significance, the population size must be extremely large, which is sometimes possible only by combining different studies—so-called meta-analyses. The problem with meta-analyses is that they compare studies that were not defined or corrected with the same parameters, which may lead to errors in interpretation. This has usually been the case with epidemiological studies of diabetes-cancer and obesity-cancer.
Neither diabetes nor cancer should be treated as single diseases. Diabetes encompasses several metabolic and hormonal disorders (such as T1DM, T2DM, or gestational diabetes) with shared features, such as hyperglycemia. Similarly, lung cancer and color cancer are two distinct diseases that cannot be analyzed together. Some diabetes-related factors, such as obesity or treatment, may greatly affect the diabetes-cancer association, but not all studies included in a meta-analysis take this into consideration.
The present review discusses current epidemiologic evidence regarding the association of obesity and diabetes with cancer, and provides an overview of the potential mechanisms implicated in such an association.
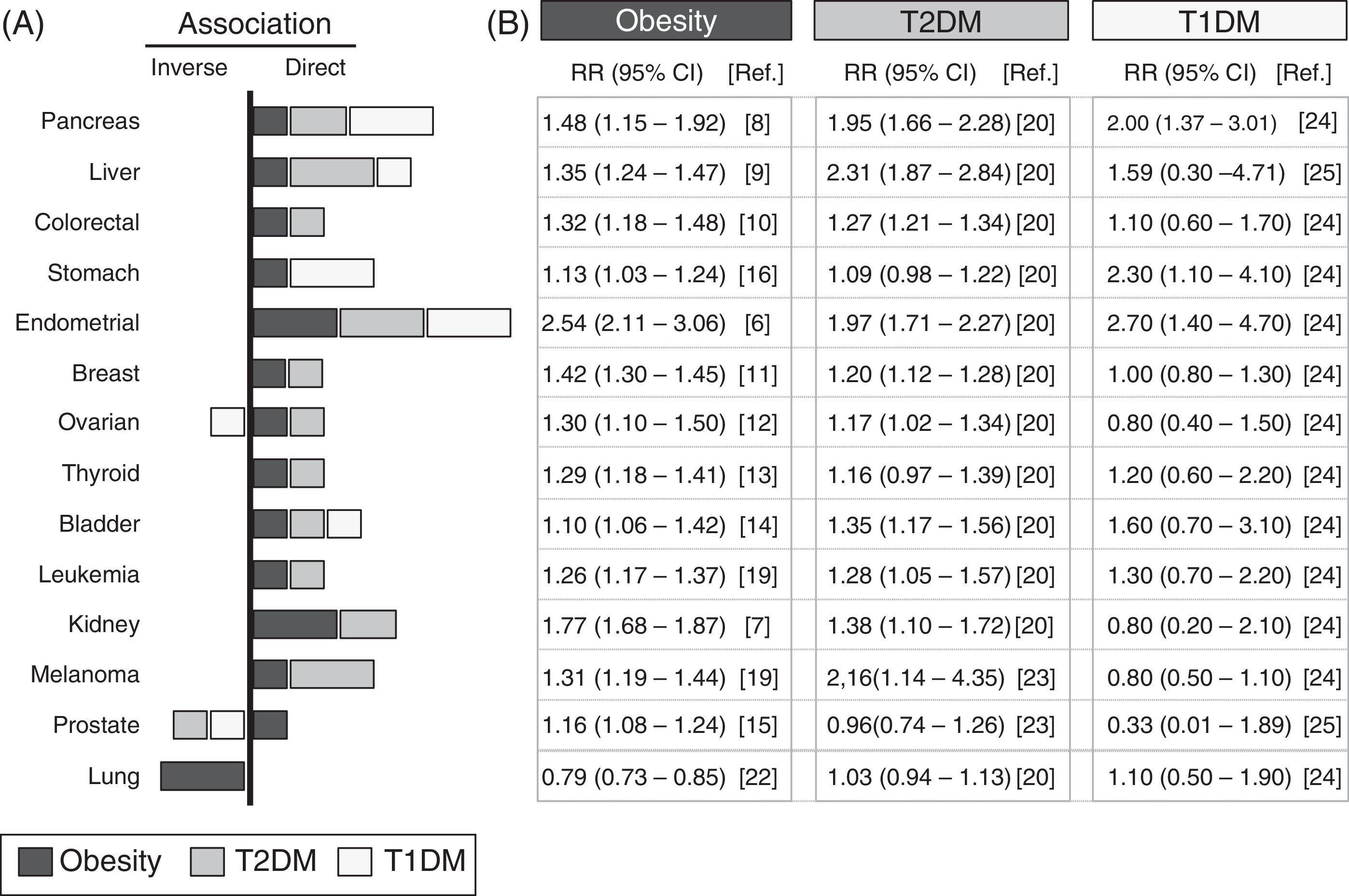
Analysis of epidemiological data linking obesity, diabetes, and cancerA systematic review is provided of epidemiological data collected from studies conducted over the past 10 years, which are summarized in Fig. 1. The meta-analyses with the most cases and which incorporated the largest number of corrections for possible confounding factors were selected. The association with different types of cancer of T1DM, T2DM and obesity—defined as the body mass index (BMI) ≥30kg/m2—was analyzed. The panel on the right details the relative risks and their statistical significance in each type of cancer for populations with obesity, T1DM, or T2DM.
 between obesity (black bars), type 2 diabetes mellitus (T2DM, gray bars), type 1 diabetes mellitus (T1DM, white bars), and different types of cancer. Data collected from meta-analyses published in the past 10 years and included in the references. (A) Graph; (B) RR and its confidence interval. The graph only represents a positive or direct association (right side) and a negative or inverse association (left side) of the RR values when the value is significant or there is a very high trend. The bar size is proportional to the increase in RR. The values of RR given for obesity were obtained for a BMI≥30kg/m2.")
Epidemiological association between obesity, diabetes, and cancer. Summary of the relative risk (RR) between obesity (black bars), type 2 diabetes mellitus (T2DM, gray bars), type 1 diabetes mellitus (T1DM, white bars), and different types of cancer. Data collected from meta-analyses published in the past 10 years and included in the references. (A) Graph; (B) RR and its confidence interval. The graph only represents a positive or direct association (right side) and a negative or inverse association (left side) of the RR values when the value is significant or there is a very high trend. The bar size is proportional to the increase in RR. The values of RR given for obesity were obtained for a BMI≥30kg/m2.
The link between obesity and cancer has long been known,4 and has been confirmed in more recent meta-analyses.5Fig. 1 shows that the obese population shows a statistically significant increase in the RR of developing the analyzed types of cancer, except for lung cancer. The increased RR is particularly important for endometrial cancer6 (up to 2.54 times as compared to the non-obese population) and kidney cancer7 (1.77 times), followed by gastrointestinal cancers (pancreas,8 liver9 and colorectal10) and, to a lesser extent, all other types (in decreasing order: breast,11 ovary,12 thyroid,13 bladder,14 prostate,15 and stomach16).
It should be noted that, as compared to the control non-obese population, obese patients have a lower RR of lung cancer (inverse association), which is one of the most common in the general population.17 These results suggest that in epidemiological studies analyzing the relationship between obesity and cancer, including patients with lung cancer, the existence of associations with less common cancers could be masked. The fact that obesity increases the risk of some cancers and decreases the risk of others clearly suggests that the association with obesity depends on the tumor's location or origin.
Another significant limitation of meta-analyses of the relationship between obesity and cancer is the heterogeneity of the criteria used to define obesity in the different studies: some use BMI values >30kg/m2, while others use central obesity defined by waist circumference, and still others the waist-hip ratio. In order to homogenize as much as possible the studies included in this review, only obesity defined as BMI≥30kg/m2 was included. Cases of overweight (BMI 25–30kg/m2) or studies using alternative definitions of obesity were not used, even though they usually reach similar conclusions.
Diabetes and cancerThe hypothesis that there is a link between diabetes and cancer is almost one hundred years old,18 and is supported by current epidemiological data.19 Because obesity is the most important factor for developing T2DM, it is not surprising that most obesity-associated cancers are also associated with T2DM20 (Fig. 1). It may thus be hypothesized that the association between diabetes and cancer is due exclusively to the obesity present in most diabetic patients. In order to explore this hypothesis, the latest studies take into account whether or not patients are obese (or also have other possible risk factors such as smoking, sex, etc.). Results show that in patients with T2DM, the increased risk of developing gastrointestinal cancer does not change with and without obesity,21 which suggests that there are risk factors specific for T2DM but independent of obesity (Fig. 1). Other types of cancer, such as liver cancer and melanoma, which have a positive correlation with obesity, are even more closely associated with diabetes19 (Fig. 1); it may thus be possible that obesity- and diabetes-specific factors are additive. Independence of the contributions from obesity and diabetes is also demonstrated by the absence of an association between T2DM and lung cancer,20 which is inversely related to obesity.22 In this case, the potential protection provided by obesity disappears upon the development of T2DM (such as, for instance, hyperinsulinemia), but the reason for this is unknown. The decreased risk of lung cancer seen may also be offset by diabetes-specific factors (treatments and other factors) that increase RR. Although more research is needed on the possible causes, the absence of an association between lung cancer and T1DM supports the notion that there are no diabetes-specific risk factors associated with lung cancer. Perhaps the example that best illustrates the existence of independent factors in obesity and diabetes is prostate cancer, which has a positive correlation with obesity15 and an inverse correlation with T2DM.20,23 Since most cases of T2DM develop from prior obesity, T2DM-specific protective factors dominate over obesity-specific factors. The possibility that treatments for T2DM have protective effects against prostate cancer is a compelling idea that should be clarified in future research.19
Taken together, the available data allow us to conclude that obesity and diabetes contribute independently to cancer. Contributions may be positive or negative, and when obesity and diabetes concur, they can have additive, synergistic, or opposing effects, depending on cancer location and origin.
T2DM accounts for more than 90% of diagnosed cases of diabetes; thus, studies that do not distinguish between T1DM and T2DM (T1DM/T2DM) follow an association pattern similar to those that analyze data from T2DM patients only.19
The most recent epidemiological studies, particularly some meta-analyses, allow for the elucidation of the specific association between T1DM and cancer. Because of the lower incidence of T1DM, there have been fewer epidemiological studies regarding this condition, and they study small populations; many data are therefore not significant and/or inconclusive (Fig. 1). The risk of developing pancreatic, stomach, and endometrial cancer is more than two times greater in patients with T1DM24 as compared to the control population. Patients with T1DM have higher rates than the control population of gastrointestinal, some hematologic, and bladder cancer,24 and the same risk of developing colorectal, breast, kidney, and lung cancer. By contrast, they have a lower risk of prostate25 and ovarian cancer and melanoma24 (Fig. 1). These data suggest that the contribution of T1DM to the onset/progression of cancer may also be positive or negative depending on the melanoma's location/origin.
The association of T1DM and T2DM with liver, pancreatic, endometrial, bladder, and lung cancer (Fig. 1) is similar (direct or inverse), which suggests that there are common mechanisms at work. The lower risk of liver and breast cancer (and perhaps melanoma) seen in patients with T1DM as compared to T2DM may suggest that in T2DM, there are specific contributions from T1DM and from obesity. On the other hand, stomach cancer, which is not associated with T2DM, has a strong association with T1DM,24 which suggests that in this case there are also specific contributions from each type of diabetes.
An additional disadvantage of many epidemiological studies is that the diabetes-cancer relationship is bidirectional. Some types of cancer (such as some pancreatic cancers) may induce in their early stages a diabetes which is usually detected before the cancer itself and acts as an indicator that helps in early treatment.26 It should be noted that the coexistence of diabetes and cancer increases mortality even in types of cancer with an inverse association.21
Diabetes and cancer treatmentsThe potential association between antidiabetic treatments and the incidence of cancer has been highly controversial.19 In 2009, the journal Diabetología published a series of studies that related insulin treatment to cancer incidence.27,28 Retrospective studies confirmed that patients with T2DM treated with insulin had a greater incidence of breast and pancreatic cancer.29 Randomized clinical trials designed to examine other outcomes, such as retinopathy (the DIGAMY study) or cardiovascular disease (the ORIGIN study), reported conflicting results regarding the association between insulin treatments and cancer.29 Moreover, other therapies that increase circulating insulin levels, such as sulfonylureas or incretin-based treatments, do not show a clear relationship with the types of cancer associated with insulin treatment. It is possible that the circulating insulin levels induced by these therapies are not as high as those achieved after exogenous insulin administration, which could then act through related receptors. A more thorough review of the epidemiological data linking antidiabetic treatments and cancer may be found in García-Jiménez et al.19 and the included references. Despite the conflicting results, there has not been sufficient research in this area. The most common error in studies that conclude that there is no association between insulin treatments and the development of cancer is that they analyze the overall incidence of cancer, grouping diabetes-associated cancers with those not associated with DM (but which are more frequent) and with cancers with an inverse association.
Contrary to what occurs with insulin, widely used antidiabetic treatments aimed at lowering circulating glucose levels, such as metformin, have been associated in retrospective studies and in some randomized clinical trials with a decreased incidence of specific cancers, such as pancreatic, liver, colorectal, and stomach cancer.19
On the other hand, many anticancer treatments contribute to the development of diabetes (not including the effects of glucocorticoids, widely used as adjuvants19). Two anticancer therapies associated with the development of grade 3–4 hyperglycemia (>250mg/dL) deserve special attention. These therapies are intended to inhibit tyrosine kinases or the protein kinase target of rapamycin (mTOR), such as everolimus and temsirolimus.30 Insulin receptors (IR) and IGF-1 receptors (IGF-1R) have intrinsic tyrosine kinase activity, and signal through mTOR to stimulate carbohydrate metabolism, protein synthesis, and cell growth (Fig. 2). As insulin is required for most glucose cell uptake, the inhibition of insulin receptors leads directly to hyperglycemia. mTOR inhibition has been shown to decrease insulin secretion and to induce peripheral insulin resistance.31 Overall, these data suggest that the mechanisms involved in the development of hyperglycemia in response to both therapies may be related. Since cancer treatments are defined based on the molecular classification of the cancer to be treated, it is surprising that no epidemiological studies have been conducted on the association between diabetes and cancer classified according to its molecular signature. Future studies based on these criteria may possibly shed more light.
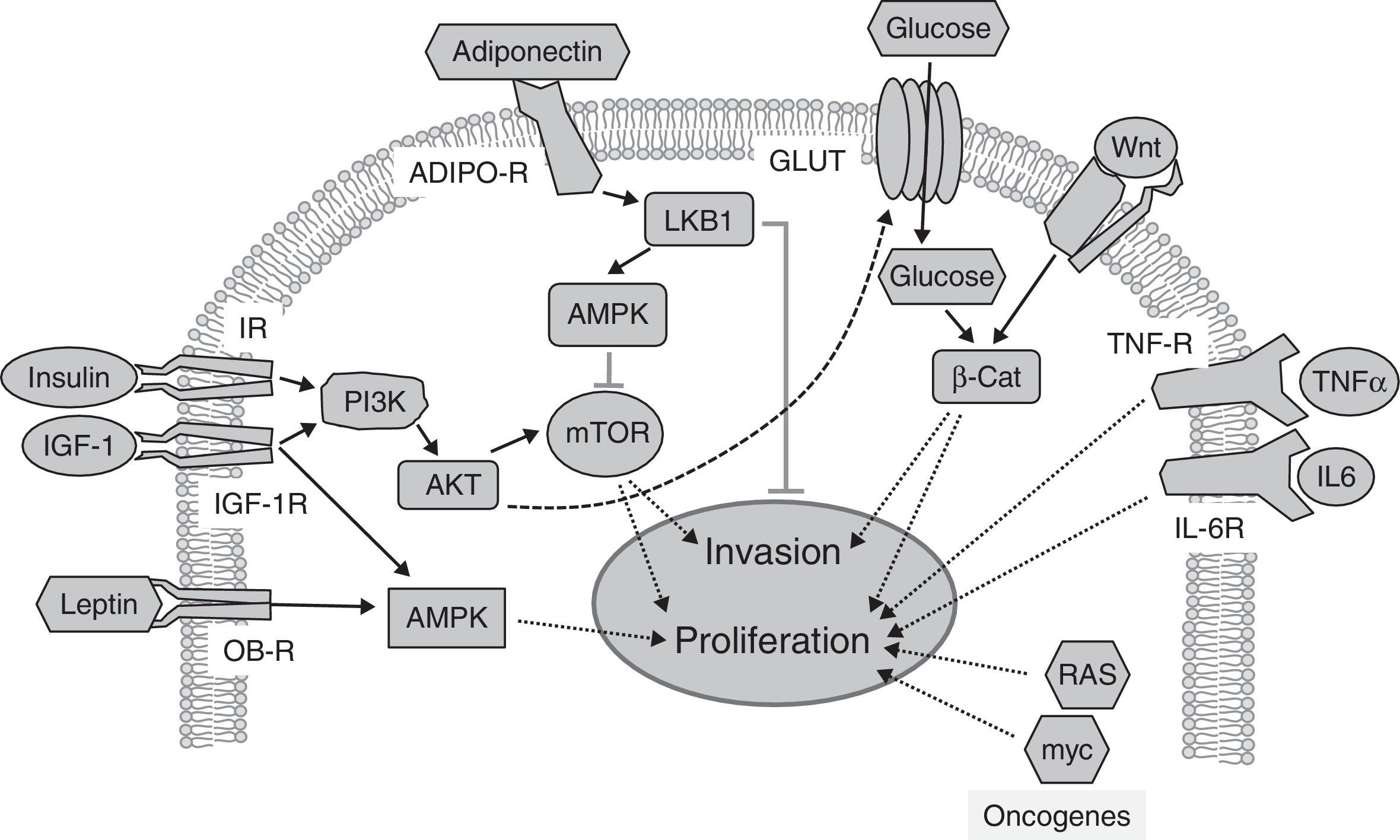
, activating the PI3K/AKT/mTOR pathway, whose targets promote tumor proliferation and invasion. Adiponectin binds to its receptors (ADIPO-R1 and ADIPO-R2), inducing the LKB1/AMPK pathway, which inhibits mTOR by inhibiting tumor proliferation and metastasis. The insulin/PI3K/AKT pathway increases glucose uptake through glucose transporters (GLUT). High glucose levels potentiate Wnt/β-Catenin signaling, inducing tumor proliferation and invasion. Circulating leptin binds to its receptor (OB-R), activating the MAPK pathway, which increases proliferation. Through their receptors IL-6R and TNF-R, interleukin-6 (IL-6) and the tumor necrosis factor-α (TNFα) activate the JAK/STAT/NF-kB pathway, which inhibits apoptosis and promotes proliferation and metastasis. Oncogenic proteins such as RAS and myc alter the expression of metabolic enzymes, increasing glycolysis, which supports increased tumor cell proliferation.")
Signaling pathways implicated in the relationship between obesity, diabetes, and cancer. Insulin and IGF-1 bind to their receptors (IR and IGF-1R respectively), activating the PI3K/AKT/mTOR pathway, whose targets promote tumor proliferation and invasion. Adiponectin binds to its receptors (ADIPO-R1 and ADIPO-R2), inducing the LKB1/AMPK pathway, which inhibits mTOR by inhibiting tumor proliferation and metastasis. The insulin/PI3K/AKT pathway increases glucose uptake through glucose transporters (GLUT). High glucose levels potentiate Wnt/β-Catenin signaling, inducing tumor proliferation and invasion. Circulating leptin binds to its receptor (OB-R), activating the MAPK pathway, which increases proliferation. Through their receptors IL-6R and TNF-R, interleukin-6 (IL-6) and the tumor necrosis factor-α (TNFα) activate the JAK/STAT/NF-kB pathway, which inhibits apoptosis and promotes proliferation and metastasis. Oncogenic proteins such as RAS and myc alter the expression of metabolic enzymes, increasing glycolysis, which supports increased tumor cell proliferation.
To sum up, epidemiology has demonstrated the existence of links between obesity, diabetes, and cancer that may help us to elucidate the molecular mechanisms that support them. However, larger-scale, randomized, long-term, well-designed studies in which cases are also broken down based on their molecular and clinical aspects (treatment type, duration and dosage of specific drugs used) are required.
Mechanisms implicated in the obesity-diabetes-cancer linkAs compared to epidemiological studies, mechanistic studies are rare, and have therefore not yielded conclusive results. Common hormonal, immunological, or metabolic abnormalities have been proposed as mechanisms linking obesity, diabetes, and cancer, but their individual roles and their interactions remain unclear.
Hormonal changesInsulin/IGF-1 axisSince the first correlations between diabetes and cancer were established, scientific debate has been dominated by the hypothesis that hyperinsulinemia (caused by a pre-diabetic condition or by the treatment itself) and the resultant increase in the bioavailability of IGF-1 may be responsible for the diabetes-cancer link.32 This hypothesis is based mainly on the fact that insulin and IGF-1 have direct33 and indirect mitogenic effects that increase both hepatic expression34 and the bioavailability of IGF-1.32 Thus, the delayed growth of tumors grafted onto animals with T1DM35 was attributed to lack of insulin. It was subsequently shown that tumor cells overexpress IR and IGF-1R,36 and that the addition of insulin to tumor cells increased proliferation by 20%–40%36; this apparently supported the hypothesis that the lack of insulin delayed tumor growth in animals with T1DM. However, each of these arguments may be refuted: delayed tumor growth in mice with T1DM may be due to immune disorders associated with immune system hyperactivity rather than to the lack of insulin or to defects in the IGF-1 axis present in T1DM37; on the other hand, tumor cells proliferate at a faster rate when insulin is added, but only in the presence of high glucose levels, on which they depend. Moreover, overexpression of IR and IGF-1R in tumor cells is not specific for the types of cancer associated with diabetes,29 as most IR is in the nucleus, where it does not usually respond to insulin.
If increased insulin and IGF-1 levels were the cause of the increased incidence of cancer in diabetic patients, IGF-1R antibodies would have a strong anticancer effect. However, the results from clinical trials have been conflicting and discouraging,38 although this could be explained by the existence of mutations in components of the signaling pathway downstream of the receptor (Fig. 2) that result in constitutive activation of the pathway. On the other hand, patients with T1DM have decreased circulating IGF-1 levels,37 despite which the incidence of stomach, pancreatic, or endometrial cancer is much higher than in the control or T2DM populations. In addition, the elevated circulating IGF-1 levels found in prostate cancer39 (inversely associated with diabetes) also challenge the value of this hypothesis. Additional evidence against this hypothesis includes tumor growth retardation in animals after insulin treatment,40 or in patient induced hypoglycemic coma.41 These results should be studied thoroughly to help us understand the mechanisms involved.
Taken together, these data suggest that increased circulating insulin levels are a factor that may contribute to the association between diabetes and some types of cancer, while they may be protective against other types.19 In any case, it is clear that high insulin levels cannot be the only factor mediating the diabetes-cancer link.
Leptin/adiponectinThe link between obesity and cancer has been attributed, amongst other factors, to an impaired secretion pattern of some adipokines such as leptin and adiponectin42 (Fig. 2). Leptin levels are usually increased in obesity.42 These changes in adipose secretome remain in diabetes, and may therefore play a causative role in the association of obesity and diabetes with certain cancers. In addition to regulating energy metabolism in the hypothalamus,43 leptin also stimulates cell growth, migration, and invasion44in vitro45 and in vivo.46 The mechanisms whereby leptin stimulates tumor growth are not clear, and may be indirect. For instance, leptin increases the production of proinflammatory cytokines in macrophages, thus stimulating cancer cells.47 Leptin also stimulates tumor growth in in vitro models of breast and colorectal cancer, activating aromatase and, consequently, estrogen synthesis.45In vivo, leptin-deficient (ob/ob) or leptin-resistant (db/db) mice do not develop breast cancer,46 which confirms the importance of leptin for tumor development.
Overall, the data suggest that leptin may contribute to the development and progression of certain types of cancer, but the mechanisms should be studied more thoroughly.
Abnormally low adiponectin levels in obesity and diabetes contrast with increased leptin levels, and have also been associated with tumor development.48 The activation of adiponectin receptors limits the proliferation of prostate, breast, and esophagus tumor cells in vitro,49 but conflicting results have been reported for other types of cancer.49,50 Human studies support the antiproliferative role of adiponectin.2 Decreased serum adiponectin levels correlate with the risk, stage, and grade of colorectal cancer.51 Low adiponectin levels also correlate with a higher incidence of breast cancer in postmenopausal women,52 and with endometrial cancer in premenopausal women.49 The antitumor effect of adiponectin has been related to the stimulation of a kinase known to inhibit the development of metastasis: the tumor suppressor LKB1.53 LKB1 phosphorylates and stimulates the AMP-activated protein kinase (AMPK), a metabolic cell sensor responsible for adapting cell growth to the availability of nutrients and growth factors (Fig. 2). AMPK (induced by the adiponectin/LKB1 pathway) phosphorylates and inhibits the activity of mTOR,48 a mediator of the effects of insulin and IGF-1. The targets of mTOR control important cancer processes such as proliferation and invasiveness (Fig. 2). The potential antitumor role of adiponectin is also supported by epidemiological studies that show an association of genetic variants of adiponectin (ADIPOQ) and its receptors (AdipoR1/R2) with the risk of developing different types of cancer.49
To sum up, hormonal changes related with the insulin/IGF-1 and leptin/adiponectin axes may contribute directly and indirectly, and independently, to the association of obesity and diabetes with cancer (Fig. 2). The interactions of these hormones in different tissues or organs with local factors such as estrogens or testosterone could undoubtedly explain some of the differences seen in some organ-specific types of cancer.
Inflammation and immunityAnother shared aspect of the pathophysiology of obesity and diabetes is a diffuse and chronic inflammatory state characterized by the increased production by adipose tissue of proinflammatory cytokines such as interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α), which leads to the development of insulin resistance.54 Increases in circulating proinflammatory cytokines induced by obesity may contribute to the development of cancer; high IL-6 and TNF-α levels have been reported in patients with different types of cancer.55 Inflammatory cytokines signal through protein kinases such as MAPK or JAK/STAT, and contribute to the biology of cancer by increasing the proliferation, survival, and accumulation of mutations56 in tumor cells. Systemically, they contribute to suppress the antitumor immunity of the host.57 However, although it seems clear that inflammatory cytokines play a significant role in tumor growth and invasiveness, the inflammation response also has antitumor activity.58,59 Thus, additional research is needed on the contribution of the inflammatory state characteristic of diabetes to tumor biology. Different immune changes are found in T1DM, as it is an autoimmune disease characterized by hyperactivity against beta cells mediated by T cells and activated antigen-presenting cells.60 Immune system hyperactivity may contribute to the inverse association with ovarian and prostate cancer, or to the lack of an association between T1DM and cancers associated with T2DM. By contrast, in types of cancer such as those of the pancreas or stomach, showing a greater association with T1DM than with the other conditions, the contribution of immune changes may possibly be lower, although other changes may be responsible for the association.
Patients with diabetes have more infections, and more complications of infections, than non-diabetics.61 Moreover, some roles of the humoral immune system (the secretion of some cytokines and complement activation) are decreased in these patients. Such immunosuppressant mechanisms decrease immune surveillance of tumor cells, which may represent another possible point of interaction between diabetes and cancer.
Metabolic changesMetabolic changes, such as hyperglycemia, represent an additional link between diabetes and cancer. High circulating glucose levels promote tumor growth through direct and indirect mechanisms.29 Glucose uptake is increased in tumor cells because of the metabolic adaptations they experience.62 The high glycolytic flux quickly produces intermediaries for proliferation at the expense of a low energy output, and to maintain it, the overexpression of glucose transporters such as GLUT1 and GLUT4 is required. Glucose also indirectly induces tumor growth by increasing circulating levels of growth factors (insulin/IGF-1), epidermal growth factor (EGF), and inflammatory cytokines.34,55
Glucose may have an impact on tumor biology by increasing circulating insulin levels. However, in tumor cells cultured in vitro, glucose promotes growth independently of insulin.63In vivo, hyperglycemic, insulin-deficient mice develop more and larger liver tumors than controls that respond to insulin.64
Other mechanisms by which glucose favors tumor biology independently of insulin include: a) the induction of increases in growth factors such as EGF in cancer cells29; b) increased cell invasiveness and migration65; and c) increases in reactive oxygen species and glycosylated products.66 Tumor cells that obtain energy through oxidative phosphorylation with high glucose availability increase the production of reactive oxygen species that favor the development of mutations (but also cell death due to the accumulation of DNA damage). The metabolic stress imposed by high glucose levels induces defects in the DNA repair mechanisms, leading to the selection of the mutations in oncogenes and tumor suppressors that promote proliferation.67 The relationship between glucose metabolism and oncogenes is bidirectional: high glucose levels induce mutations in oncogenes, and oncogenes (including RAS and myc) alter the expression of metabolic enzymes, forcing cell metabolism toward glycolysis, thus reducing oxidative damage.68 Oncogene mutations may regulate glucose consumption, determining the metabolic phenotype of each cell within a single tumor, which allows for coexistence and cooperation between cells with a different metabolism and phenotype within the tumor.
High glucose levels also have immunosuppressive effects mediated by the competitive inhibition by glucose of ascorbic acid transport to immune cells.29
A little-studied aspect of the direct actions of glucose on tumor cells is the modulation of the signaling that sustains tumor growth; one of the most important is signaling by Wnt proteins. High glucose levels amplify tumor signaling through the Wnt pathway, inducing nuclear retention of the final effector, β-catenin.63 β-Catenin is a potent transcriptional co-activator that induces the expression of proliferation, survival and invasiveness genes. The effects of glucose on β-catenin are independent of insulin, adipokines, and inflammatory cytokines.63 Other signaling pathways associated with tumor proliferation and modulated by high glucose levels are the MAPK and AKT kinase pathways,69 which mediate the cell effects of insulin.
Taken together, these data suggest that hyperglycemia is a risk factor independent of hormonal and inflammatory factors for the development of cancer in diabetic patients.
In recent years, the results from cancer research have emphasized the importance of immunity and metabolism, and oncometabolites have even been found.70 Overall, the emerging concept is that certain metabolites modulate signaling in the tumor cell, inducing the adaptations required to support all tumor characteristics.
Conclusions and future perspectives- •
Epidemiological data have established direct or inverse relationships of obesity, T1DM and T2DM with the incidence of certain types of cancer depending on their location. Studies based on the molecular signature of tumors are needed to clarify these relationships. Future epidemiological studies should unify the correction factors used and the methods to measure them. The standardization of blood glucose levels using HbA1c values, and the use of the BMI and waist circumference values to define obesity are proposed.
- •
Obesity, T2DM, and T1DM contribute with independent mechanisms (metabolic, hormonal, and immune) to the incidence of tissue-specific cancers. Studies that identify such mechanisms will facilitate future epidemiological studies that delimit the contribution of each disease to the incidence of cancer. Mechanistic studies are also needed to identify new therapeutic targets.
- •
The relationships between diabetes and cancer are reciprocal: Cancer may develop from diabetes, and diabetes from cancer, and the coexistence of the diseases significantly worsens prognosis. Studies are needed for further understanding of the development of diabetes during the early course of some cancers.
- •
Some antidiabetic treatments, such as insulin, have a positive association with the development of some types of cancer, while others, such as metformin, are associated with a decreased risk of some cancers. However, epidemiological studies that take into consideration such important controls as treatment duration and drug dose are needed.
- •
Some anticancer agents contribute to the development of diabetes, and specific, industry-independent studies that take into consideration dose and time, and may help in selecting the most adequate treatment for each patient are needed.
This study was supported by Instituto de Salud Carlos III (FIS) (PI12/01201 for A.V., and PI13/01150 for C.G.-J.), MINECO/FEDER SAF2015-69964-R for A.V., Universidad Rey Juan Carlos-Banco de Santander (Grupo Excelencia QUINANOAP), the European Union (PIEF-GA-2013-626098 for A.C.-C.), and the European Molecular Biology Organization (EMBO) (ALTF 800-2013 for A.C.-C.).
Conflicts of interestThe authors state that they have no conflicts of interest.
Please cite this article as: Gutiérrez-Salmerón M, Chocarro-Calvo A, García-Martínez JM, de la Vieja A, García-Jiménez C. Bases epidemiológicas y mecanismos moleculares implicados en las asociaciones de obesidad y diabetes con cáncer. Endocrinol Diabetes Nutr. 2017;64:109-117.
