




Hace casi un siglo que se hipotetizó la asociación entre la diabetes y el cáncer. Hoy, numerosos estudios epidemiológicos sostienen que las poblaciones con obesidad y/o diabetes poseen una mayor predisposición a padecer cáncer en órganos específicos. Los mecanismos moleculares subyacentes se desconocen. Las alteraciones metabólicas, hormonales e inmunológicas que comparten la obesidad, la diabetes y el cáncer pueden contribuir a justificar la relación existente. Por otra parte, la influencia de los tratamientos antidiabéticos en la aparición/evolución de algunos cánceres y la inducción de la diabetes por los tratamientos antineoplásicos han despertado una gran controversia debido a las implicaciones éticas y los intereses comerciales asociados. Esta actualización de los datos epidemiológicos presenta un enfoque mecanístico que sugiere la existencia de múltiples mecanismos comunes y diferenciales que asocian la obesidad y la diabetes tipo1 y tipo2 a ciertos cánceres. Identificar los mecanismos responsables de la asociación diabetes-cáncer es un reto de la investigación actual; la clasificación de los cáncer por sus firmas moleculares podría facilitar futuros estudios mecanísticos y epidemiológicos.
The association between diabetes and cancer was hypothesized almost one century ago. Today, a vast number of epidemiological studies support that obese and diabetic populations are more likely to experience tissue-specific cancers, but the underlying molecular mechanisms remain unknown. Obesity, diabetes, and cancer share many hormonal, immune, and metabolic changes that may account for the relationship between diabetes and cancer. In addition, antidiabetic treatments may have an impact on the occurrence and course of some cancers. Moreover, some anticancer treatments may induce diabetes. These observations aroused a great controversy because of the ethical implications and the associated commercial interests. We report an epidemiological update from a mechanistic perspective that suggests the existence of many common and differential individual mechanisms linking obesity and type 1 and 2 diabetes mellitus to certain cancers. The challenge today is to identify the molecular links responsible for this association. Classification of cancers by their molecular signatures may facilitate future mechanistic and epidemiological studies.
La obesidad, la diabetes y el cáncer son enfermedades metabólicas. La obesidad predispone a padecer diabetes, y ambas (obesidad y diabetes) representan factores de riesgo para muchos tipos de cáncer1. Según la Organización Mundial de la Salud (OMS), el 39% de la población mundial adulta tiene sobrepeso y más del 13% presenta obesidad, cifras que se han duplicado desde 1980. El 9% de la población adulta padece diabetes, y se estima que este valor se duplicará para el año 2030 (http://apps.who.int/iris/bitstream/10665/148114/1/9789241564854_eng.pdf). De igual forma, la OMS prevé que habrá un aumento de aproximadamente un 70% de casos de cáncer en 2030 (http://publications.iarc.fr/Non-Series-Publications/World-Cancer-Reports/World-Cancer-Report-2014).
Tanto la diabetes tipo1 (DM1) como la diabetes tipo2 (DM2) comparten con el cáncer alteraciones de tipo hormonal (defectos en los ejes insulina/IGF-1 y leptina/adiponectina)2, metabólico (afectan a los hidratos de carbono y lípidos) e inmunológico (aumentos de citoquinas inflamatorias circulantes)3.
Los estudios epidemiológicos en salud humana tratan de asesorar sobre el impacto en la salud de la población de patologías, factores sociales, medioambientales, etc. Una manera de medir dicho impacto es estimar el aumento o la disminución del riesgo relativo (RR) de sufrir otra patología. Para ello se necesitan grandes grupos poblacionales de casos y controles (prospectivos o retrospectivos), que tienen la ventaja de que los resultados pueden generalizarse al conjunto poblacional. El inconveniente es que para obtener una significación estadística del estudio, el tamaño poblacional debe ser excesivamente grande, lo que en ocasiones solo se consigue con la combinación de distintos estudios, los llamados metaanálisis. El problema de los metaanálisis es que se comparan estudios que no han sido definidos ni corregidos con los mismos parámetros y pueden conducir a errores de interpretación. Este ha sido el caso habitual en los estudios epidemiológicos entre diabetes-cáncer y obesidad-cáncer.
Ni la diabetes ni el cáncer deben tratarse como una enfermedad única. La diabetes engloba distintos trastornos metabólicos y hormonales (como la DM1, la DM2 o la diabetes gestacional) con características comunes, como la hiperglucemia. De la misma manera, el cáncer de pulmón y el cáncer de colon son dos enfermedades distintas que no pueden analizarse conjuntamente. Algunos factores relacionados con la diabetes, como la obesidad o los tratamientos, pueden influir notablemente en la asociación diabetes-cáncer, y no todos los estudios de un metaanálisis los tienen en cuenta.
En la presente revisión discutimos las evidencias epidemiológicas actuales sobre la asociación entre la obesidad y la diabetes con el cáncer y ofrecemos una visión general de los posibles mecanismos implicados en dicha asociación.
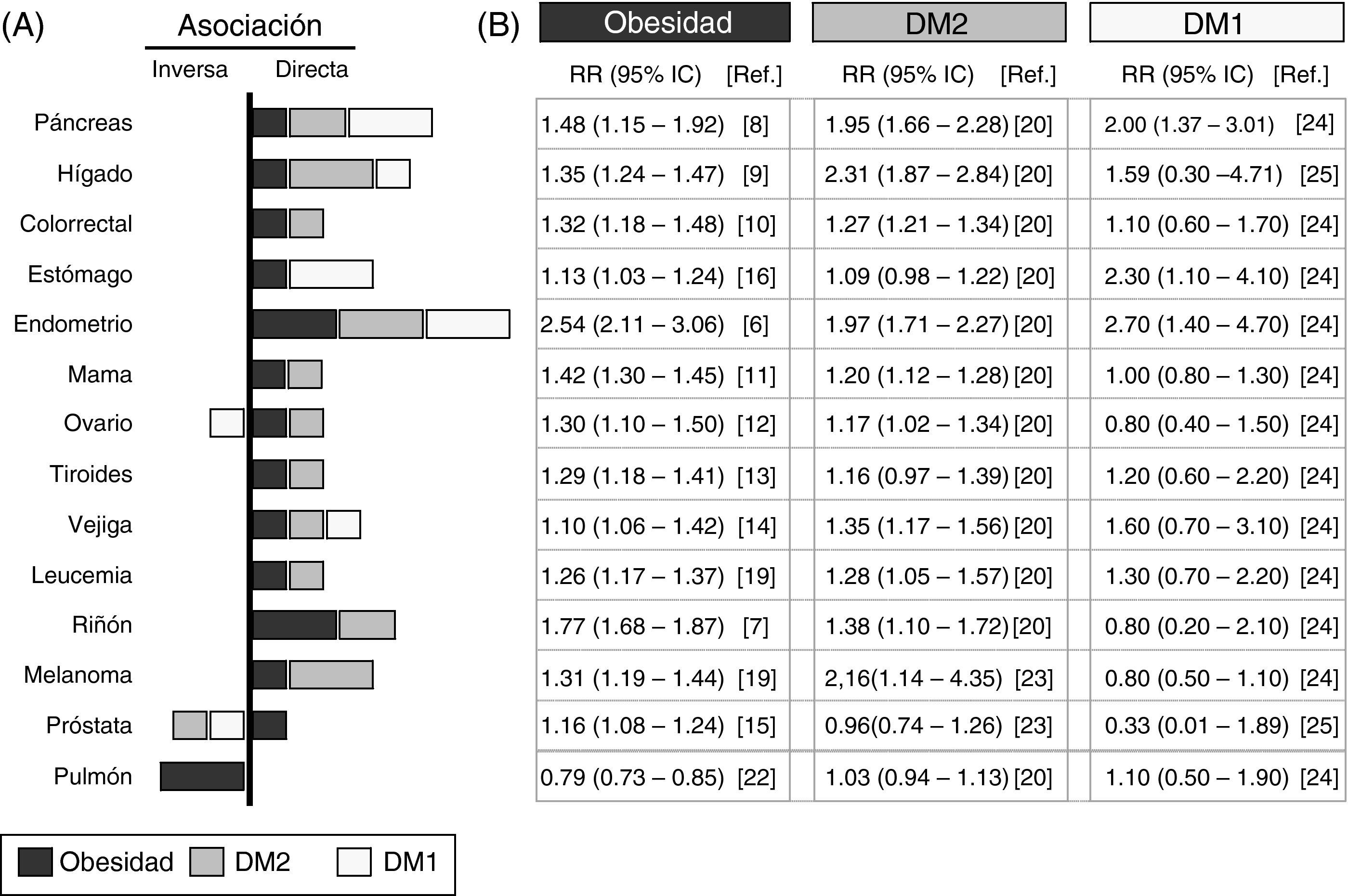
Análisis de los datos epidemiológicos que asocian la obesidad, la diabetes y el cáncerPresentamos una revisión sistemática de los datos epidemiológicos recogidos en estudios de los últimos 10años y resumidos en la figura 1. Hemos seleccionado los metaanálisis con mayor número de casos y que han introducido el mayor número de correcciones con posibles factores de confusión. Se ha analizado la asociación con distintos tipos de cáncer de la DM1, la DM2 y la obesidad, definida con un índice de masa corporal (IMC) ≥30kg/m2. El panel de la derecha detalla los riesgos relativos y su significación estadística en cada tipo de cáncer para las poblaciones con la obesidad, con la DM2 o con la DM1.
 entre la obesidad (cuadrados negros), la diabetes tipo2 (DM2, cuadrados grises), la diabetes tipo1 (DM1, cuadrados blancos) y los distintos tipos de cáncer. Datos obtenidos de meta-análisis publicados en los últimos 10años e indicados en las referencias que se incluyen. A)gráfica; B)RR y su intervalo de confianza. En la gráfica solo se representa la asociación positiva o directa (lado derecho) y negativa o inversa (lado izquierdo) de los valores de RR cuando el valor es significativo o la tendencia es muy alta. El tamaño de la barra es proporcional al aumento del RR. Los valores de RR indicados para obesidad fueron obtenidos para un IMC≥30kg/m2.")
Asociación epidemiológica entre la obesidad, la diabetes y el cáncer. Resumen del riesgo relativo (RR) entre la obesidad (cuadrados negros), la diabetes tipo2 (DM2, cuadrados grises), la diabetes tipo1 (DM1, cuadrados blancos) y los distintos tipos de cáncer. Datos obtenidos de meta-análisis publicados en los últimos 10años e indicados en las referencias que se incluyen. A)gráfica; B)RR y su intervalo de confianza. En la gráfica solo se representa la asociación positiva o directa (lado derecho) y negativa o inversa (lado izquierdo) de los valores de RR cuando el valor es significativo o la tendencia es muy alta. El tamaño de la barra es proporcional al aumento del RR. Los valores de RR indicados para obesidad fueron obtenidos para un IMC≥30kg/m2.
La asociación entre obesidad y cáncer se conoce desde hace tiempo4, y se confirma en los metaanálisis más recientes5. La figura 1 muestra que la población con obesidad tiene un aumento estadísticamente significativo del RR de padecer los cánceres analizados, excepto el cáncer de pulmón. El aumento del RR es especialmente importante para el cáncer de endometrio6 (hasta 2,54 veces respecto a la población no obesa) y de riñón7 (1,77 veces), seguido por cánceres gastrointestinales (páncreas8, hígado9 y colorrectales10) y en menor medida el resto (por orden decreciente: mama11, ovario12, tiroides13, vejiga14, próstata15 y estómago16).
Llama la atención que los pacientes con obesidad tienen un RR inferior de sufrir cáncer de pulmón que la población control sin obesidad (asociación inversa), siendo este uno de los tipos más frecuentes de cáncer en la población general17. Estos resultados evidencian que en los estudios epidemiológicos que analizan la relación obesidad-cáncer, incluyendo pacientes con cáncer de pulmón, se podría enmascarar la existencia de asociaciones con otros cánceres menos frecuentes. El hecho de que la obesidad aumente el riesgo de algunos cánceres y lo disminuya en otros indica claramente que la asociación con obesidad depende de la localización o del origen del tumor.
Otra limitación importante de los metaanálisis que han analizado la relación obesidad-cáncer es la heterogeneidad de los criterios utilizados para definir la obesidad entre los diferentes estudios, utilizando unos el IMC>30kg/m2, otros la obesidad central medida con la circunferencia de la cintura, y otros mediante el índice cintura/cadera. En este trabajo y con el fin homogeneizar al máximo los estudios incluidos, solo hemos considerado la obesidad definida con IMC≥30kg/m2; no se consideraron los casos de sobrepeso (IMC de 25-30kg/m2) ni estudios que valoran la obesidad con definiciones alternativas, aunque generalmente coinciden en las conclusiones.
Diabetes y cáncerLa hipótesis de una asociación diabetes-cáncer tiene casi un siglo18, y los datos epidemiológicos actuales la respaldan19. Siendo la obesidad el factor de riesgo más importante para padecer DM2, no es sorprendente que la mayoría de los cánceres asociados a la obesidad estén también asociados a la DM220 (fig. 1). Podría plantearse entonces que la asociación encontrada entre la diabetes y el cáncer se debe exclusivamente a la obesidad que presentan la mayor parte de los pacientes con diabetes. Para explorar esta hipótesis, los estudios más recientes tienen en cuenta si los pacientes presentan obesidad (además de otros posibles factores de riesgo como hábito de fumador, sexo, etc.). Los resultados muestran que en pacientes con DM2 el riesgo aumentado de sufrir cáncer gastrointestinal no varía tengan o no obesidad21, evidenciando la existencia de factores de riesgo propios de la DM2 e independientes de la obesidad (fig. 1). Otros tipos de cáncer, como el hepático o el melanoma, que se asocian positivamente a la obesidad, se asocian aún más positivamente a la diabetes19 (fig. 1), por lo que podrían estar sumándose factores propios de la obesidad y de la diabetes. La independencia de las contribuciones de la obesidad y la diabetes se pone también de manifiesto por la falta de asociación entre la DM2 y el cáncer de pulmón20, asociado de forma inversa a la obesidad22. En este caso, la posible protección derivada de la obesidad desaparece al desarrollar DM2 (como, por ejemplo, la hiperinsulinemia), pero se desconoce su origen. Es posible también que la disminución del riesgo de cáncer de pulmón observada con la obesidad quede compensada por factores exclusivos de la diabetes (tratamientos u otros) que incrementen el RR. Aunque es necesaria una investigación profunda de las posibles causas, la falta de asociación de cáncer de pulmón con DM1 refuerza la idea de que no hay factores de riesgo propios de la diabetes que la asocien a cáncer de pulmón. Quizás el ejemplo que mejor ilustra la existencia de factores independientes en la obesidad y la diabetes es el cáncer de próstata, asociado positivamente a la obesidad15 e inversamente con la DM220,23. Dado que la mayoría de los casos de DM2 se desarrollan a partir de una obesidad previa, los factores protectores propios de la DM2 son dominantes sobre los factores de riesgo propios de la obesidad. La posibilidad de que los tratamientos contra la DM2 ejerzan efectos protectores en cáncer de próstata constituye un punto muy interesante que ha de esclarecerse en investigaciones futuras19.
Tomando en conjunto todos los datos, se concluye que se ejercen contribuciones independientes al desarrollo de cáncer desde la obesidad y la diabetes; dichas contribuciones pueden ser positivas o negativas, y cuando hay obesidad y diabetes pueden relacionarse de formas aditivas, sinérgicas u opuestas, dependiendo de la localización o del origen del cáncer.
La DM2 representa más del 90% de las diabetes diagnosticadas y, en consecuencia, los estudios que no distinguen entre la DM1 y la DM2 (DM1/2) siguen un patrón de asociación similar a aquellos que solo analizan datos de pacientes con DM219.
Los estudios epidemiológicos más recientes, y en especial algunos metaanálisis, están permitiendo aclarar la asociación específica de la DM1 con el cáncer. Dada la menor incidencia de la DM1, los estudios epidemiológicos son menos numerosos e incluyen poblaciones pequeñas, por lo que muchos datos no son significativos y/o concluyentes todavía (fig. 1). El riesgo de sufrir cáncer de páncreas, estómago y endometrio es más de dos veces mayor en pacientes con DM124 que en la población control. La población con DM1 presenta frecuencias aumentadas de cánceres gastrointestinales, algunos hematológicos o de vejiga24, mientras que presenta el mismo riesgo que la población control de sufrir cáncer colorrectal, de mama, de riñón y de pulmón. En contraste, destaca el menor riesgo de sufrir cáncer de próstata25, de ovario y, quizás, melanoma24 (fig. 1). Estos datos indican que la contribución de la DM1 al inicio/progresión tumoral puede ser también positiva o negativa dependiendo de la localización/origen del tumor de melanoma.
Si comparamos la asociación de la DM1 y la DM2 con cánceres de hígado, páncreas, endometrio, vejiga y pulmón (fig. 1), es similar (directa o inversa), sugiriendo la existencia de mecanismos comunes. El menor riesgo de cáncer de hígado, de mama (y quizás melanoma) observado en pacientes con DM1 en comparación con pacientes con DM2 podría indicar que en la DM2 se suman contribuciones comunes con la DM1 y con la obesidad. Por otra parte, los cánceres de estómago, que no se asocian con la DM2, presentan una fuerte asociación con la DM124, indicando la existencia, también en este caso, de contribuciones propias de cada tipo de diabetes.
Un inconveniente añadido de muchos estudios epidemiológicos es que las relaciones diabetes-cáncer son bidireccionales. Ciertos tipos de cáncer (como algunos de páncreas) pueden generar una diabetes en los estadios iniciales, que suele ser detectada antes que el propio cáncer, actuando como un indicador que ayuda al tratamiento temprano26. Cabe destacar que la coexistencia de la diabetes con el cáncer aumenta la mortalidad incluso en tipos de cáncer que se asocian de forma inversa21.
Tratamientos en diabetes y cáncerLa posible asociación entre los tratamientos antidiabéticos con la incidencia de cáncer ha generado una gran controversia19. En 2009 la revista Diabetología publicó una serie de estudios que relacionaban el tratamiento con insulina con la incidencia de cáncer27,28. Los estudios retrospectivos confirmaron que los pacientes con DM2 tratados con insulina presentaban mayor incidencia de cáncer de mama y de páncreas29. El análisis de ensayos clínicos aleatorizados, diseñados para estudiar otros resultados como la retinopatía (estudio DIGAMY) o la enfermedad cardiovascular (estudio ORIGIN), arrojaron resultados contradictorios sobre la asociación de los tratamientos con insulina y cáncer29. Además, otras terapias que conducen a aumentar la insulina circulante, como las sulfonilureas o los tratamientos basados en incretinas, no muestran una clara relación con los tipos de cáncer asociados al tratamiento con insulina. Quizá las concentraciones de insulina circulante inducidas por estos tratamientos son menos elevadas que las alcanzadas por administración exógena de insulina, que podría entonces actuar a través de receptores relacionados. Una revisión más exhaustiva sobre los datos epidemiológicos que asocian los tratamientos antidiabéticos a cánceres puede consultarse en García-Jiménez et al.19 y referencias incluidas. A pesar de los resultados contradictorios, no se ha impulsado convenientemente la investigación en el área, y el error más repetido en los estudios que concluyen una falta de asociación entre los tratamientos con insulina y el desarrollo de cáncer es que analizan la incidencia de cáncer global, agrupando cánceres asociados a diabetes junto con los no asociados pero más frecuentes, y con los asociados inversamente.
En sentido opuesto a lo que ocurre con la insulina, los tratamientos antidiabéticos dirigidos a reducir la glucosa circulante y ampliamente utilizados, como la metformina, se han asociado en estudios retrospectivos y algunos ensayos clínicos aleatorizados con disminución de incidencia de cánceres específicos, como los de páncreas, hígado, colorrectal y estómago19.
Por otro lado, muchos tratamientos antineoplásicos contribuyen al desarrollo de diabetes (sin incluir los efectos de los glucocorticoides, ampliamente usados como adyuvantes19). Especial atención merecen dos terapias antineoplásicas que se asocian con el desarrollo de hiperglucemia de grado 3-4 (> 250mg/dl). Estas terapias van dirigidas a inhibir tirosín-quinasas o bien a la proteína-quinasa diana de rapamicina (mTOR), como everolimus y temsirolimus30. Los receptores de insulina (IR) e IGF-1 (IGF-1R) tienen actividad tirosín-quinasa intrínseca y señalizan a través de mTOR para estimular el metabolismo glucídico, la síntesis proteica y el crecimiento celular (fig. 2). Siendo la insulina necesaria para la captación celular de la mayor parte de la glucosa, la inhibición de los receptores de insulina conduce directamente a hiperglucemia. La inhibición de mTOR se ha demostrado que disminuye la secreción de insulina e induce resistencia periférica a insulina31. En conjunto, estos datos sugieren que los mecanismos involucrados en el desarrollo de la hiperglucemia en respuesta a ambas terapias podrían estar relacionados. Sabiendo que los tratamientos oncológicos se definen en base a la clasificación molecular del cáncer a tratar, llama la atención que no se hayan hecho estudios epidemiológicos sobre la asociación entre diabetes y cáncer clasificando los cánceres por sus firmas moleculares. Quizá futuros estudios puedan arrojar más luz basándose en estos criterios.
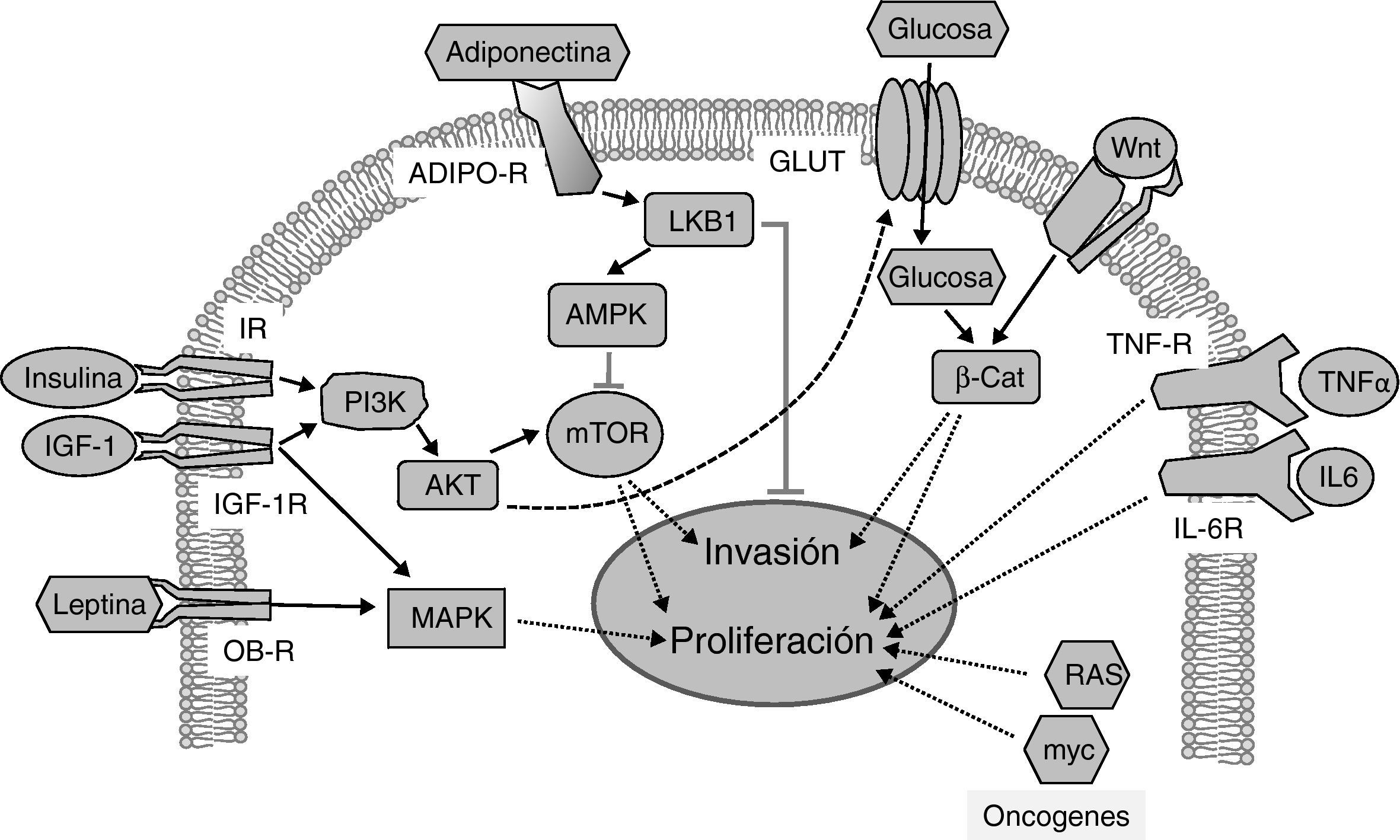
 activando la vía PI3K/AKT/mTOR, cuyas dianas promueven proliferación e invasión tumoral. La adiponectina se une a sus receptores (ADIPO-R1 y ADIPO-R2) induciendo la vía LKB1/AMPK, que inhibe a mTOR inhibiendo la proliferación tumoral y metástasis. La vía insulina/PI3K/AKT aumenta la captura de glucosa a través de sus transportadores (GLUT). Las elevadas concentraciones de glucosa potencian la señalización por Wnt/β-Catenina induciendo proliferación e invasión tumoral. La leptina circulante se une a su receptor (OB-R) activando la vía de MAPK que aumenta proliferación. La interleucina 6 (IL-6) y el factor de necrosis tumoral-α (TNFα) a través de sus receptores IL-6R y TNF-R activan la vía JAK/STAT/NF-kB, que inhibe apoptosis y promueve la proliferación y la metástasis. Proteínas oncogénicas como RAS y myc alteran la expresión de enzimas metabólicas aumentando la glucólisis que sustenta la proliferación aumentada en las células tumorales.")
Vías de señalización implicadas en la relación entre la obesidad, la diabetes y el cáncer. La insulina y el IGF-1 se unen a sus receptores (IR e IGF-1R, respectivamente) activando la vía PI3K/AKT/mTOR, cuyas dianas promueven proliferación e invasión tumoral. La adiponectina se une a sus receptores (ADIPO-R1 y ADIPO-R2) induciendo la vía LKB1/AMPK, que inhibe a mTOR inhibiendo la proliferación tumoral y metástasis. La vía insulina/PI3K/AKT aumenta la captura de glucosa a través de sus transportadores (GLUT). Las elevadas concentraciones de glucosa potencian la señalización por Wnt/β-Catenina induciendo proliferación e invasión tumoral. La leptina circulante se une a su receptor (OB-R) activando la vía de MAPK que aumenta proliferación. La interleucina 6 (IL-6) y el factor de necrosis tumoral-α (TNFα) a través de sus receptores IL-6R y TNF-R activan la vía JAK/STAT/NF-kB, que inhibe apoptosis y promueve la proliferación y la metástasis. Proteínas oncogénicas como RAS y myc alteran la expresión de enzimas metabólicas aumentando la glucólisis que sustenta la proliferación aumentada en las células tumorales.
En definitiva, la epidemiología está poniendo de manifiesto la existencia de conexiones entre la obesidad, la diabetes y el cáncer que podrían ayudar a esclarecer los mecanismos moleculares que las sustentan. Sin embargo, se necesitan estudios a mayor escala, aleatorizados, con un tiempo de seguimiento largo y que estén bien diseñados, desglosando los casos también por aspectos moleculares y clínicos (tipo de tratamiento, tiempo y dosis de los medicamentos específicos empleados).
Mecanismos implicados en la asociación obesidad-diabetes-cáncerComparados con los estudios epidemiológicos, los estudios mecanísticos son escasos, por lo que no han arrojado resultados concluyentes. Se han propuesto las alteraciones hormonales, inmunológicas o metabólicas comunes como mecanismos que conectan la obesidad, la diabetes y el cáncer sin haber esclarecido el papel de cada una ni sus interacciones.
Alteraciones hormonalesEje insulina/IGF-1Desde que se establecieron las primeras correlaciones entre la diabetes y el cáncer, la hipótesis de que la hiperinsulinemia (derivada del estado pre-diabético o del propio tratamiento) y el subsiguiente aumento en la biodisponibilidad de IGF-1 pudieran ser responsables de la asociación diabetes-cáncer ha dominado el escenario científico32. Esta hipótesis se basa fundamentalmente en que la insulina y el IGF-1 ejercen efectos mitogénicos directos33 e indirectos, aumentando tanto la expresión hepática de IGF-134 como su biodisponibilidad32. Así, el retardo en el crecimiento de tumores injertados a animales con DM135 se atribuyó a la falta de insulina. Posteriormente se demostró que las células tumorales sobreexpresan IR e IGF-1R36, y que añadir insulina a las células tumorales aumentaba un 20-40% la proliferación36, lo que parecía confirmar la hipótesis de que la falta de insulina retardaba el crecimiento de tumores en animales con DM1. Sin embargo, cada uno de estos argumentos puede ser rebatido: el retardo en el crecimiento tumoral en ratones con DM1 se puede deber a las anomalías inmunológicas relacionadas con la hiperreactividad del sistema inmune en vez de a la falta de insulina o a los defectos en el eje de IGF-1 presentes en DM137; por otra parte, las células tumorales proliferan en mayor medida al añadir insulina únicamente si hay altas concentraciones de glucosa, de la que dependen. Además, la sobreexpresión de IR e IGF-1R en células tumorales no es específica de los tipos tumorales asociados a diabetes29, encontrándose la mayor parte del IR en el núcleo donde difícilmente responderá a insulina.
Si los aumentos en los niveles de la insulina e IGF-1 fueran la causa de la incidencia aumentada de cáncer en los pacientes con diabetes, los anticuerpos contra IGF-1R deberían tener un potente efecto antineoplásico. Sin embargo, los resultados de los ensayos clínicos han sido contradictorios y poco alentadores38, aunque ello podría explicarse por la existencia de mutaciones en componentes de la vía de señalización por debajo del receptor (fig. 2) que resultasen en la activación constitutiva de la vía. Por otra parte, los pacientes con DM1 tienen niveles circulantes de IGF-1 disminuidos37 y, sin embargo, la incidencia de cánceres de estómago, páncreas o endometrio es mucho más elevada que en la población control o con DM2. Además, los niveles elevados de IGF-1 circulante encontrados en cáncer de próstata39, un cáncer inversamente asociado a diabetes, también cuestionan el valor de esta hipótesis. Otras evidencias en contra de esta hipótesis son el retraso en el crecimiento de tumores en animales tras el tratamiento con insulina40 o en pacientes en los que se indujo un coma hipoglucémico41. Estos resultados merecen explorarse en profundidad para entender los mecanismos implicados.
En conjunto, estos datos apuntan a que los niveles aumentados de insulina circulante representan un factor que puede contribuir a la asociación entre diabetes y algunos tipos de cáncer, mientras en otros podría suponer un factor protector19. En cualquier caso queda claro que los elevados niveles de insulina no pueden ser el único factor que media la relación diabetes-cáncer.
Leptina/adiponectinaLa asociación entre la obesidad y el cáncer se ha atribuido entre otros factores a la aparición de un perfil alterado de secreción de ciertas adipoquinas, como la leptina y la adiponectina42 (fig. 2). Los niveles de leptina suelen estar aumentados en la obesidad42. Puesto que estas alteraciones del secretoma adiposo permanecen durante la diabetes, podría atribuírseles un papel causal en la asociación de la obesidad y la diabetes con ciertos cánceres. La leptina, además de regular el metabolismo energético en el hipotálamo43, también estimula el crecimiento, la migración y la invasión celular44in vitro45 e in vivo46. Los mecanismos por los que la leptina estimula el crecimiento tumoral no están claros y pueden ser indirectos. Por ejemplo, la leptina aumenta la producción de citoquinas proinflamatorias por los macrófagos, estimulando así a las células cancerosas47. Además, la leptina estimula el crecimiento tumoral en modelos in vitro de cáncer de mama y colorrectales, activando la aromatasa y, en consecuencia, la síntesis de estrógenos45. In vivo, ratones deficientes en leptina (ob/ob) o resistentes a leptina (db/db) no desarrollan tumores de mama46, confirmando la importancia de la leptina en el desarrollo tumoral.
En conjunto, los datos apuntan a que la leptina podría contribuir al desarrollo y progresión tumoral en ciertos tipos de cáncer, aunque los mecanismos deben ser explorados en mayor profundidad.
Los niveles anormalmente bajos de adiponectina durante la obesidad y la diabetes contrastan con los aumentos en leptina y también se han asociado al desarrollo tumoral48. La activación de los receptores de adiponectina limita la proliferación de células tumorales de próstata, de mama y de esófago in vitro49, aunque se han publicado resultados contradictorios para otros tipos de cáncer49,50. Los estudios en humanos confirman el papel antiproliferativo de la adiponectina2. Los niveles séricos disminuidos de adiponectina se han correlacionado con el riesgo, el estadio y el grado del cáncer colorrectal51. También, los niveles bajos de adiponectina en mujeres posmenopáusicas se correlacionan con incidencia aumentada de cáncer de mama52, y en mujeres premenopáusicas, con cáncer de endometrio49. El efecto antitumoral de la adiponectina se ha relacionado con la estimulación de una quinasa conocida por inhibir el desarrollo de metástasis: el supresor tumoral LKB153. LKB1 fosforila y estimula a la proteína quinasa activada por AMP: AMPK, un sensor metabólico celular responsable de adaptar el crecimiento celular a la disponibilidad de nutrientes y factores de crecimiento (fig. 2). La AMPK (inducida por la vía adiponectina/LKB1) fosforila e inhibe la actividad de mTOR48, mediador de los efectos de la insulina e IGF-1. Las dianas de mTOR controlan procesos tan importantes en cáncer como la proliferación y la invasividad (fig. 2). El posible papel antitumoral de la adiponectina viene también apoyado por estudios epidemiológicos que demuestran una asociación entre variantes genéticas de la adiponectina (ADIPOQ) y sus receptores (AdipoR1/R2) con el riesgo de padecer diferentes tipos de cáncer49.
En suma, las alteraciones hormonales relacionadas con los ejes de la insulina/IGF-1 y la leptina/adiponectina pueden contribuir directa e indirectamente y de modo independiente a la asociación entre la obesidad y la diabetes con el cáncer (fig. 2). Sin duda, las interacciones de estas hormonas en distintos tejidos u órganos con factores locales como estrógenos o testosterona podrían explicar algunas de las diferencias observadas en cánceres específicos de ciertos órganos.
Estado inflamatorio e inmunidadOtro aspecto compartido de la fisiopatología de la obesidad y la diabetes es un estado inflamatorio difuso y crónico, caracterizado por aumentos en la producción de citoquinas proinflamatorias tales como la interleucina 6 (IL-6) y el factor de necrosis tumoral alfa (TNF-α) por el tejido adiposo, lo cual conduce al desarrollo de resistencia a la insulina54. Los aumentos en citoquinas proinflamatorias circulantes inducidos por la obesidad pueden contribuir al desarrollo de cáncer; se han descrito niveles elevados de IL-6 y TNF-α en pacientes con distintos tipos de cáncer55. Las citoquinas inflamatorias señalizan a través de proteínas quinasas como las MAPK o JAK/STAT y contribuyen a la biología del cáncer aumentando la proliferación, la supervivencia y la acumulación de mutaciones56 en las células tumorales; a nivel sistémico contribuyen a la supresión de la inmunidad antitumoral del huésped57. Sin embargo, aunque parece claro que las citoquinas inflamatorias juegan un papel importante en el crecimiento y la invasividad del tumor, la respuesta inflamatoria también tiene actividad antitumoral58,59, por lo que la contribución del estado inflamatorio característico de la diabetes a la biología tumoral todavía necesita investigarse. La DM1 presenta alteraciones inmunológicas diferentes al ser una enfermedad autoinmune caracterizada por la hiperreactividad contra las células beta mediada por linfocitosT y células presentadoras de antígeno activadas60. Una hiperreactividad del sistema inmune podría contribuir a la asociación inversa con cánceres de ovario y próstata, o a la falta de asociación entre la DM1 y cánceres asociados a la DM2. Por el contrario, en cánceres como los de páncreas o estómago, que están más asociados a DM1 que a las otras patologías, quizá la contribución de las alteraciones inmunes sea menor, aunque otras alteraciones pudieran ser responsables de la asociación.
Los pacientes diabéticos tienen más infecciones y con más complicaciones que los pacientes no diabéticos61. Además, algunas funciones del sistema inmune humoral (secreción de ciertas citoquinas y activación del sistema de complemento) están disminuidas en estos pacientes. Dichos mecanismos inmunosupresores disminuyen la vigilancia inmunológica frente a las células tumorales, representando otro posible punto de interacción entre diabetes y cáncer.
Alteraciones metabólicasLas alteraciones metabólicas, como la hiperglucemia, representan otra conexión entre la diabetes y el cáncer. Los niveles elevados de glucosa circulante favorecen el crecimiento tumoral a través de mecanismos directos e indirectos29. La captura de glucosa está incrementada en las células tumorales62 como consecuencia de las adaptaciones metabólicas que sufren. El elevado flujo glucolítico genera rápidamente intermediarios para la proliferación a costa de un bajo rendimiento energético y su mantenimiento requiere la sobreexpresión de transportadores de glucosa como GLUT1 y GLUT4. La glucosa también induce el crecimiento tumoral de manera indirecta aumentando los niveles circulantes de factores de crecimiento (insulina/IGF-1), factor de crecimiento epidérmico (EGF) y de citoquinas inflamatorias34,55.
La influencia de la glucosa en la biología tumoral puede ejercerse al inducir aumentos en la insulina circulante. Sin embargo, la glucosa favorece el crecimiento independientemente de la insulina en células tumorales cultivadas in vitro63. In vivo, los ratones hiperglucémicos deficientes en insulina desarrollan más tumores de hígado y más grandes que sus controles con respuesta a insulina64.
Otros mecanismos por los que la glucosa favorece la biología tumoral independientemente de la insulina incluyen: a)la inducción de aumentos de factores de crecimiento como el EGF en células de cáncer29; b)un incremento en la invasividad y la migración celular65, y c) aumentos en especies reactivas de oxígeno y productos glicosilados66. Las células tumorales que obtengan su energía mediante la fosforilación oxidativa con alta disponibilidad de glucosa aumentarán la producción de especies reactivas de oxígeno que favorecen la aparición de mutaciones (aunque también la muerte celular por acumulación de daños en el ADN). El estrés metabólico impuesto por los niveles elevados de glucosa induce defectos en los mecanismos de reparación del ADN, seleccionándose las mutaciones en oncogenes y supresores tumorales que favorecen la proliferación67. Las relaciones entre el metabolismo de la glucosa y los oncogenes son bidireccionales: altas concentraciones de glucosa inducen mutaciones en oncogenes, y los oncogenes (incluyendo a RAS y myc) alteran la expresión de enzimas metabólicas, forzando el metabolismo celular hacia glucólisis y reduciendo así los daños oxidativos68. Las mutaciones oncogénicas pueden regular el consumo de glucosa determinando el fenotipo metabólico de cada célula dentro de un mismo tumor, permitiendo la coexistencia y cooperación entre células con distinto metabolismo y fenotipo dentro del tumor.
Los niveles elevados de glucosa también ejercen efectos inmunosupresores mediados por la inhibición competitiva por glucosa del transporte de ácido ascórbico a las células del sistema inmune29.
Un aspecto poco explorado de las acciones directas de la glucosa sobre la célula tumoral es la modulación de la señalización que sostiene el crecimiento tumoral; una de las más importantes es la señalización por proteínas Wnt. Las concentraciones elevadas de glucosa amplifican la señalización tumoral de la vía Wnt induciendo la retención nuclear de su efector final, la β-catenina63. La β-catenina es un potente co-activador transcripcional que induce la expresión de genes de proliferación, supervivencia e invasividad. Los efectos de la glucosa sobre la β-catenina son independientes de insulina, adipoquinas o citoquinas de inflamación63. Otras vías de señalización relacionadas con proliferación tumoral y moduladas por elevadas concentraciones de glucosa son las vías de las quinasas MAPK y AKT69, que median los efectos celulares de la insulina.
Tomando en conjunto todos estos datos, puede concluirse que la hiperglucemia representa un factor de riesgo independiente a los factores hormonales y los inflamatorios para el desarrollo de cáncer en pacientes diabéticos.
En los últimos años los resultados de la investigación en cáncer han destacado la importancia de la inmunidad y del metabolismo con el hallazgo incluso de oncometabolitos70. En general, el concepto emergente es que ciertos metabolitos modulan la señalización en la célula tumoral induciendo las adaptaciones necesarias para sostener todas las características tumorales.
Conclusiones y perspectivas futuras- •
Los datos epidemiológicos establecen asociaciones (directas o inversas) de la obesidad, la DM1 o la DM2 con la incidencia de ciertos cánceres según su localización. Se necesitan estudios basados en la firma molecular de los tumores para clarificar estas relaciones. Los futuros estudios epidemiológicos deben unificar los factores de corrección incluidos y la manera de medirlos. Proponemos la normalización de los valores de glucemia utilizando los valores de HbA1c y de obesidad con el IMC y circunferencia de la cintura.
- •
La obesidad, la DM2 y la DM1 contribuyen con mecanismos independientes (metabólicos, hormonales e inmunes) a la incidencia de cánceres tejido-específicos. Los estudios que identifiquen dichos mecanismos facilitarán futuros estudios epidemiológicos que delimiten la aportación de cada patología a la incidencia de cánceres. Los estudios mecanísticos son también necesarios para identificar nuevas dianas terapéuticas.
- •
Las relaciones entre la diabetes y el cáncer son recíprocas: puede desarrollarse cáncer a partir de la diabetes o diabetes a partir del cáncer, y la coexistencia de ambas enfermedades agrava mucho el pronóstico. Se necesitan estudios para entender el desarrollo de diabetes durante la evolución temprana de algunos cánceres.
- •
Algunos tratamientos antidiabéticos, como la insulina, se asocian positivamente al desarrollo de algunos cánceres, y otros, como la metformina, a la disminución del riesgo de ciertos cánceres. Sin embargo, se necesitan estudios epidemiológicos donde se tengan en cuenta controles tan importantes como el tiempo de tratamiento y la dosis del fármaco.
- •
Algunos antineoplásicos contribuyen al desarrollo de diabetes, siendo necesarios estudios específicos, independientes de la industria y que tengan en cuenta la dosis y el tiempo que puedan ayudar a decidir las terapias más adecuadas para cada paciente.
El presente trabajo ha sido financiado por el Instituto de Salud CarlosIII (FIS) (PI12/01201 para A.V. y PI13/01150 para C.G.-J.), MINECO/FEDER SAF2015-69964-R para A.V., la Universidad Rey Juan Carlos-Banco de Santander (Grupo Excelencia QUINANOAP), la Comunidad Económica Europea (PIEF-GA-2013-626098 para A.C.-C.) y la Organización Europea de Biología Molecular (EMBO) (ALTF 800-2013 para A.C.-C.).
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
