




Primary hyperparathyroidism (PHPT) is the leading cause of hypercalcemia. Although the most common presentation is sporadic PHPT attributable to a single adenoma, there are inherited forms in 10% of the cases.1 The most common inherited variants form part of syndromes in which PHPT is associated with other endocrine disorders, including multiple endocrine neoplasia (MEN) type 1 and 2, hyperparathyroidism-jaw tumor syndrome (HPT-JT), familial hypocalciuric hypercalcemia (FHH), and familial isolated hyperparathyroidism (FIHP).
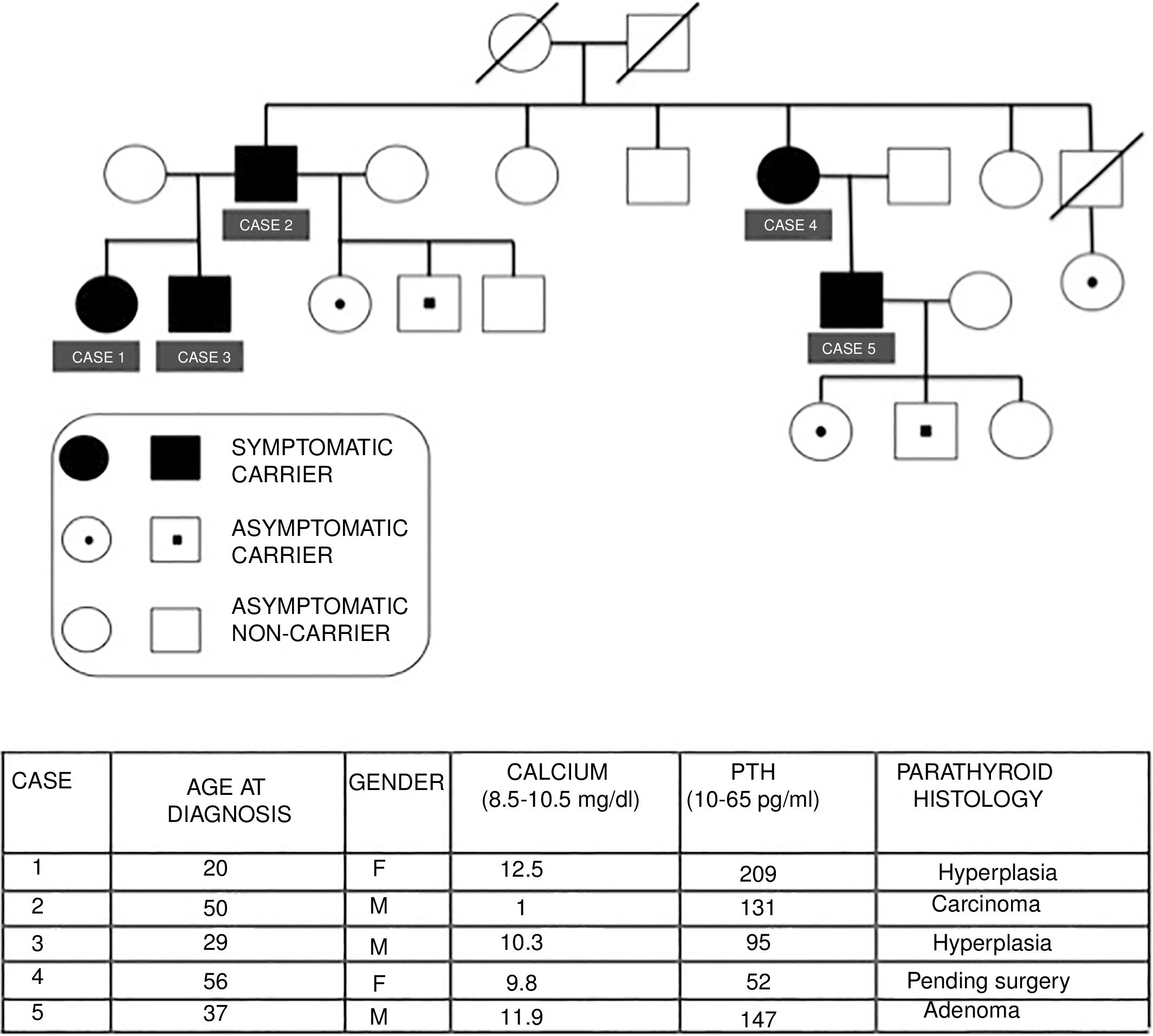
We present the cases of a family with FIHP, in which the index case (case 1) was first evaluated in 2006. Since then a total of three generations have been studied, comprising 15 family members, of which 10 are carriers of the HRPT2 gene mutation, while 5 are symptomatic carriers. Fig. 1 describes the family tree and summarizes the main clinical, laboratory, and histological characteristics of the affected patients.

Case 1: A 20-year-old female consulted due to PHPT with nephrolithiasis and osteopenia. The MIBI scintigraphic findings proved negative on two occasions. A right inferior parathyroidectomy was performed, with intraoperative reporting of parathyroid hyperplasia. The intraoperative parathyroid hormone (PTH) levels decreased adequately according to the Miami criteria.2 After surgery, normal PTH and calcium levels were maintained.
Case 2: A 50-year-old male presented with symptoms of nephrolithiasis. The laboratory tests revealed hypercalcemia, hypophosphatemia and inappropriate PTH elevation, with negative MIBI scintigraphy. There was no evidence of associated endocrine disorders. Surgery revealed an enlarged left superior parathyroid gland. The other glands showed no alterations. A left superior parathyroidectomy was performed, and a lowering of the PTH levels was verified intraoperatively. The histology report indicated parathyroid carcinoma. Subsequent controls proved adequate.
On identifying two members with PHPT in the same family, a molecular study was made including an analysis of the HRPT2 gene (1q25-q31) in peripheral blood through automated sequencing, using specific DNA primers flanking the region where the mutation responsible for the disease is located in this family. This study revealed a heterozygous mutation in exon 6, c.456_459dup/p.Ala154IlefsX16, in the first two cases and in the other carriers.
Case 3: A 29-year-old male presented with normocalcemic hyperparathyroidism. The MIBI scan revealed an ectopic parathyroid adenoma. A subtotal parathyroidectomy plus thymectomy was performed. The pathology report indicated parathyroid hyperplasia.
A genetic study was also made of the siblings of the father, and was found to be negative in 5 of them and positive in one (case 4).
Case 4: A 56-year-old female, currently under evaluation due to osteopenia, renal colic episodes and vitamin D deficiency. Completion of the study is pending, with subsequent surgery if required.
Case 5: A 37-year-old male presented with symptomatic PHPT associated with microlithiasis and osteopenia of the femur. A subtotal parathyroidectomy with thymectomy was recently carried out. The histology report indicated a right inferior parathyroid adenoma, with hyperplasia of the other glands. The patient has three offspring, two of whom carry the gene.
The asymptomatic carriers of the familial mutation undergo annual controls with laboratory tests and neck and kidney ultrasound explorations.
Familial isolated hyperparathyroidism is considered an autonomous non-syndromic entity or an incomplete expression of one of the genetic syndromes causing PHPT. The diagnosis is established through exclusion and requires the presence of at least two first-degree relatives with PHPT and no other endocrine manifestations1 such as hypophyseal and pancreatic disease characteristic of MEN-1 or fibrous tumors of the jaw and kidney in HPT-JT.3 It has not been established whether familial isolated hyperparathyroidism is a variant or an early stage of MEN-1 syndrome.
The disorder usually appears between 20 and 25 years, as in our index case, and approximately 30 years earlier than in the case of sporadic PHPT. The affected patients show multi-gland involvement, with a lower cure rate and an increased risk of recurrent PHPT and carcinomas compared with sporadic PHPT.1,4 In addition, they develop severe hypercalcemia more often than patients with MEN-1.3
The disorder exhibits an autosomal dominant hereditary trait, with the existence of no specific gene. However, germinal mutations in MEN-1, HRPT2 and CASR have been described in a significant number of families, so mutations of the same gene may be responsible for different syndromes.1,5
The clinical manifestations are severe, and the histological findings are usually consistent with adenomas or carcinomas, as in our case. Hyperplasias are less common. However, no patients showed cystic changes in the histopathological study, typical of HPT-JT.1
Recently, a germinal mutation in GCM-2 has been described in families with familial isolated hyperparathyroidism, characterized by higher PTH levels, a greater risk of multi-gland disease and carcinoma, and with a lower biochemical cure rate.6,7
On the other hand, mutation of the HRTP2 (CDC73) gene is identified in 15–20% of all sporadic parathyroid gland carcinomas. As a result, some authors4,5 consider it to be a tumor suppressor gene. Patients with newly diagnosed parathyroid carcinoma should undergo a careful review of their family history and should be offered a genetic study for HRTP2 gene mutation.3
Asymptomatic carriers require optimal prospective monitoring, including neck ultrasound and periodic serum calcium and PTH measurements, in order to ensure early detection of the disease.8
While the indication of surgery for the management of sporadic PHPT is well established, the treatment of choice in patients with familial isolated hyperparathyroidism associated with HRPT2 remains controversial. Several authors8,9 argue that in view of the high rate of recurrence or persistence of the disease, the morbidity associated with repeat surgery, and the risk of developing parathyroid carcinoma,8,10 a subtotal parathyroidectomy should become the initial strategy in patients with mutation of the HRPT2 gene. Other authors suggest that total parathyroidectomy is the best treatment strategy, even in the absence of suspected cancer.3 We advocate subtotal parathyroidectomy with thymectomy to avoid the development of parathyroid carcinomas.
In cases of PHPT in young adults, particularly when associated with a family history of PHPT and in the absence of other syndromic conditions, it is important to extend genetic studies to other mutations in addition to MEN-1, including HRPT2. In families with familial isolated hyperparathyroidism and mutation of the HRPT2 gene, close monitoring of asymptomatic carriers is crucial, and a more aggressive surgical approach is recommended in patients with PHPT because of the increased risk of developing parathyroid carcinoma.
Thanks are due to Dr. M. Robledo of the Centro Nacional de Investigaciones Oncológicas for the genetic study.
Please cite this article as: Luján D, Sánchez A, Meoro A, Albarracín A, Candel MF. Hiperparatiroidismo familiar aislado asociado al gen HRPT2. Endocrinol Diabetes Nutr. 2018;65:470–472.
