




Primary hyperparathyroidism (PHP) is a very common disease in endocrinology clinics. Its prevalence in Western countries has been estimated at 0.86%,1 and its incidence has increased notably in recent years due to more frequent measurement of blood calcium levels in the general population and more detailed study of patients with osteoporosis or kidney stones.
PHP is caused by hyperfunction of the parathyroid glands resulting in raised levels, or inappropriately high levels within the normal range, of parathyroid hormone (PTH). This excess of PTH can cause hypercalcaemia, hypophosphataemia, osteoporosis, nephrolithiasis, deterioration in glomerular filtration rate, and other complications.
The pathological cause of this parathyroid hyperfunction is an adenoma in 80–85% of cases, a diffuse hyperplasia of all the parathyroid tissue in 15–20% and parathyroid carcinoma in less than 1%.1
Most cases of PHP are sporadic, but approximately 5–10% occur as a result of germline mutations.2,3 Others are clinical syndromes which may be associated with other tumours (such as multiple endocrine neoplasia [MEN]) or isolated familial hyperparathyroidism. Some of these mutations also occur somatically in the tumour tissue of patients with sporadic PHP. However, the main clinical interest is to identify patients with germline mutations, as knowing this would allow us to:
- a)
Determine which patients are at risk of developing tumours in other locations (e.g. gastro-entero-pancreatic or thymic neuroendocrine tumours [NET], which can be life-threatening).
- b)
Carefully plan the surgical approach to PHP, as some familial forms require subtotal parathyroidectomy to minimise the risk of recurrence.
- c)
Carry out lifelong follow-up, due to the increased risk of recurrence or development of other associated tumours.
- d)
Carry out genetic screening of first-degree relatives, to allow early diagnosis of potentially serious syndromes.
The clinical management guidelines for PHP corresponding to the Fourth International Workshop3 establish that genetic testing should be considered in patients with clinical suspicion, but leaves the subject quite open. Some scientific surgical societies, such as the American Association of Endocrine Surgeons,4 give slightly more specific recommendations and advise a genetic study in patients with PHP diagnosed before the age of 40 with multiglandular disease, or in those with a family history or evidence of a related syndrome.
However, the genetic study of patients with PHP has not become sufficiently widespread, probably due to the financial cost of genetic studies and the lack of specific recommendations on who and which genes to study.
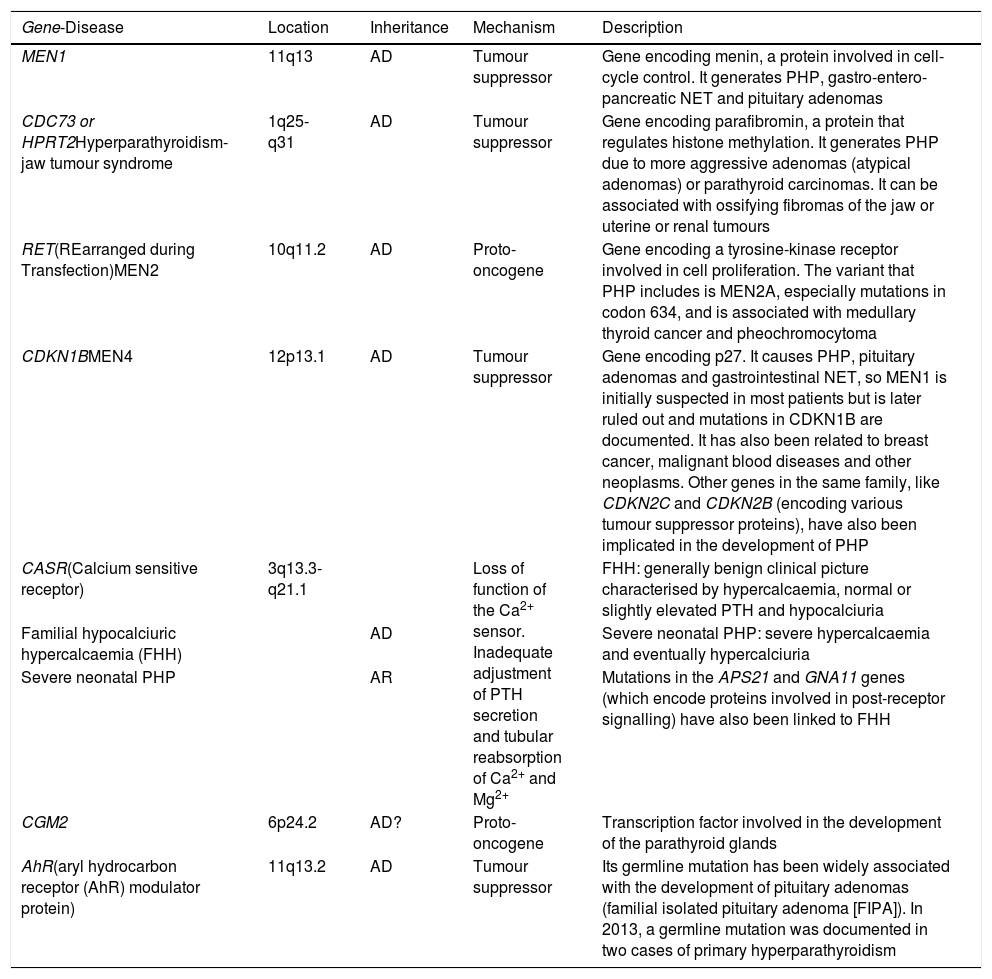
The main germline mutations and/or molecular mechanisms5–9 related to PHP are summarised in Table 1.
Main germline mutations related to primary hyperparathyroidism (PHP).
| Gene-Disease | Location | Inheritance | Mechanism | Description |
|---|---|---|---|---|
| MEN1 | 11q13 | AD | Tumour suppressor | Gene encoding menin, a protein involved in cell-cycle control. It generates PHP, gastro-entero-pancreatic NET and pituitary adenomas |
| CDC73 or HPRT2Hyperparathyroidism-jaw tumour syndrome | 1q25-q31 | AD | Tumour suppressor | Gene encoding parafibromin, a protein that regulates histone methylation. It generates PHP due to more aggressive adenomas (atypical adenomas) or parathyroid carcinomas. It can be associated with ossifying fibromas of the jaw or uterine or renal tumours |
| RET(REarranged during Transfection)MEN2 | 10q11.2 | AD | Proto-oncogene | Gene encoding a tyrosine-kinase receptor involved in cell proliferation. The variant that PHP includes is MEN2A, especially mutations in codon 634, and is associated with medullary thyroid cancer and pheochromocytoma |
| CDKN1BMEN4 | 12p13.1 | AD | Tumour suppressor | Gene encoding p27. It causes PHP, pituitary adenomas and gastrointestinal NET, so MEN1 is initially suspected in most patients but is later ruled out and mutations in CDKN1B are documented. It has also been related to breast cancer, malignant blood diseases and other neoplasms. Other genes in the same family, like CDKN2C and CDKN2B (encoding various tumour suppressor proteins), have also been implicated in the development of PHP |
| CASR(Calcium sensitive receptor) | 3q13.3-q21.1 | Loss of function of the Ca2+ sensor. Inadequate adjustment of PTH secretion and tubular reabsorption of Ca2+ and Mg2+ | FHH: generally benign clinical picture characterised by hypercalcaemia, normal or slightly elevated PTH and hypocalciuria | |
| Familial hypocalciuric hypercalcaemia (FHH) | AD | Severe neonatal PHP: severe hypercalcaemia and eventually hypercalciuria | ||
| Severe neonatal PHP | AR | Mutations in the APS21 and GNA11 genes (which encode proteins involved in post-receptor signalling) have also been linked to FHH | ||
| CGM2 | 6p24.2 | AD? | Proto-oncogene | Transcription factor involved in the development of the parathyroid glands |
| AhR(aryl hydrocarbon receptor (AhR) modulator protein) | 11q13.2 | AD | Tumour suppressor | Its germline mutation has been widely associated with the development of pituitary adenomas (familial isolated pituitary adenoma [FIPA]). In 2013, a germline mutation was documented in two cases of primary hyperparathyroidism |
AD: autosomal dominant; AR: autosomal recessive; NET: neuroendocrine tumours.
Studies of systematic mutation screening in at-risk patients with PHP have shown variable performance. In Europe, Vierimaa et al.10 analysed the MEN1, CDC73, CASR, AIP and CDKN1B genes in 29 Finnish patients with data suggestive of familial PHP (family history, onset at <40 years, multiglandular disease, other tumours suggestive of multiglandular neoplastic syndrome), only finding a mutation in MEN1 in one patient. In an American population, Starker et al.11 analysed the genes MEN1, CASR and CDC73 in 86 patients with PHP diagnosed before the age of 45 and without other syndromic data, showing deleterious mutations in eight of them (four MEN1, three CASR and one CDC73). The authors concluded that genetic testing in patients diagnosed with PHP before the age of 45 is relevant and should be routinely offered to all patients. In Japan, Mizusawa et al.12 analysed 13 families with familial hyperparathyroidism with variable phenotypes, and found three families with mutations in CDC73 and one with mutation in MEN1. They add cystic parathyroid adenomas as a risk phenotype for mutations in CDC73. Published in 2020, the largest study to date13 analysed a panel made up of the genes MEN1, CDC73, CASR, CDKN1A, CDKN1B, CDKN2B, CDKN2C, RET, GCM2, GNA11 and AP2S1 in 121 British subjects with a risk profile, and found the presence of pathogenic variants in 19 patients (16%).
Our group carried out a study in 40 subjects from Sabadell with PHP and some element of risk (age ≤45, family history, risk histology, associated tumour, multiglandular disease or recurrent PHP), analysing a gene panel composed of MEN1, RET, CDC73, CDKN1B, CDKN2B, CDKN2C, CASR, APS21, GNA11, AIP and GCM2, and identified germline mutations (pathogenic or variants of uncertain significance) in nine subjects (22%).14
We can conclude that genetic study should be implemented in clinical practice in our setting in patients with PHP in whom any degree of associated family history is suspected, as there is now more evidence available on how this screening should be carried out, and the financial cost of analysing these gene panels has reduced significantly.
Please cite this article as: Capel I, Mazarico-Altisent I, Baena N. Estudio genético en el hiperparatiroidismo primario: ¿a quién y qué genes estudiar? Endocrinol Diabetes Nutr. 2022;69:237–239.
