




El hiperparatiroidismo primario (HPP) es una enfermedad muy habitual en las consultas de endocrinología. Su prevalencia en los países occidentales se ha estimado en el 0,86%1, y su incidencia se ha incrementado de forma notable en los últimos años debido a una determinación más frecuente de la calcemia en la población general y por un estudio más detallado de los pacientes con osteoporosis o litiasis renal.
El HPP está causado por una hiperfunción de las glándulas paratiroides que genera niveles aumentados, o inapropiadamente altos dentro del rango de la normalidad, de hormona paratiroidea (PTH). Este exceso de PTH puede producir hipercalcemia, hipofosfatemia, osteoporosis, nefrolitiasis, deterioro del filtrado glomerular y otras complicaciones.
La base anatomopatológica de esta hiperfunción paratiroidea es en el 80-85% de los casos un adenoma, en el 15-20% una hiperplasia difusa de todo el tejido paratiroideo y en menos del 1% un carcinoma paratiroideo1.
La mayor parte de los casos de HPP son esporádicos, pero aproximadamente del 5 al 10% se producen como consecuencia de mutaciones en línea germinal2,3. En ocasiones se trata de síndromes clínicos que pueden asociar otros tumores (como las neoplasias endocrinas múltiples [MEN]) o tratarse de hiperparatiroidismo familiar aislado. Algunas de estas mutaciones también se dan de forma somática en el tejido tumoral de los pacientes afectos por casos esporádicos. No obstante, el interés clínico principal es identificar a los pacientes con mutaciones en línea germinal, ya que conocer este hecho permitiría:
- a)
Saber qué pacientes tienen riesgo de desarrollar tumores en otras localizaciones (p.ej., tumores neuroendocrinos [TNE] gastro-entero-pancreáticos o tímicos, que pueden generar compromiso vital).
- b)
Plantear de forma cuidadosa el abordaje quirúrgico del HPP, ya que algunas formas familiares requieren paratiroidectomía subtotal para minimizar el riesgo de recidiva.
- c)
Realizar seguimiento de por vida, por el mayor riesgo de recidiva o de aparición de otros tumores asociados.
- d)
Llevar a cabo el cribado genético de familiares de primer grado, que permitirá un diagnóstico precoz de síndromes potencialmente graves.
La guía de manejo clínico del HPP correspondiente al Fourth International Workshop3 establece que debe plantearse el estudio genético en pacientes con sospecha clínica, pero deja el tema bastante abierto. Algunas sociedades científicas quirúrgicas, como la American Association of Endocrine Surgeons4, dan recomendaciones algo más concretas y plantean estudio genético en los pacientes con HPP diagnosticado antes de los 40años con enfermedad multiglandular o en aquellos con historia familiar o datos de un síndrome relacionado.
No obstante, el estudio genético de pacientes con HPP no se ha generalizado de forma satisfactoria, probablemente por el coste económico de los estudios genéticos y por la falta de recomendaciones concretas sobre a quién y qué genes estudiar.
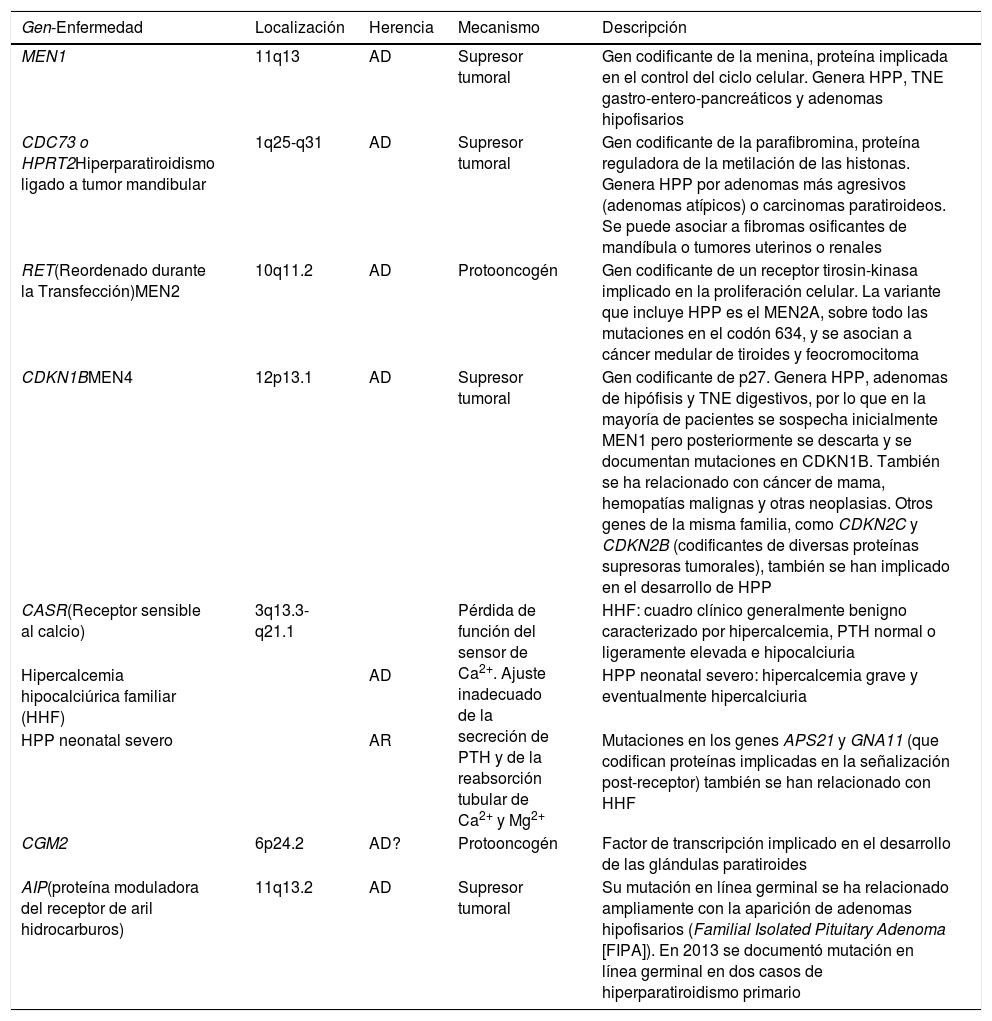
Las principales mutaciones en línea germinal y/o mecanismos moleculares5-9 relacionados con HPP se resumen en la tabla 1.
Principales mutaciones en línea germinal relacionadas con hiperparatiroidismo primario (HPP)
| Gen-Enfermedad | Localización | Herencia | Mecanismo | Descripción |
|---|---|---|---|---|
| MEN1 | 11q13 | AD | Supresor tumoral | Gen codificante de la menina, proteína implicada en el control del ciclo celular. Genera HPP, TNE gastro-entero-pancreáticos y adenomas hipofisarios |
| CDC73 o HPRT2Hiperparatiroidismo ligado a tumor mandibular | 1q25-q31 | AD | Supresor tumoral | Gen codificante de la parafibromina, proteína reguladora de la metilación de las histonas. Genera HPP por adenomas más agresivos (adenomas atípicos) o carcinomas paratiroideos. Se puede asociar a fibromas osificantes de mandíbula o tumores uterinos o renales |
| RET(Reordenado durante la Transfección)MEN2 | 10q11.2 | AD | Protooncogén | Gen codificante de un receptor tirosin-kinasa implicado en la proliferación celular. La variante que incluye HPP es el MEN2A, sobre todo las mutaciones en el codón 634, y se asocian a cáncer medular de tiroides y feocromocitoma |
| CDKN1BMEN4 | 12p13.1 | AD | Supresor tumoral | Gen codificante de p27. Genera HPP, adenomas de hipófisis y TNE digestivos, por lo que en la mayoría de pacientes se sospecha inicialmente MEN1 pero posteriormente se descarta y se documentan mutaciones en CDKN1B. También se ha relacionado con cáncer de mama, hemopatías malignas y otras neoplasias. Otros genes de la misma familia, como CDKN2C y CDKN2B (codificantes de diversas proteínas supresoras tumorales), también se han implicado en el desarrollo de HPP |
| CASR(Receptor sensible al calcio) | 3q13.3-q21.1 | Pérdida de función del sensor de Ca2+. Ajuste inadecuado de la secreción de PTH y de la reabsorción tubular de Ca2+ y Mg2+ | HHF: cuadro clínico generalmente benigno caracterizado por hipercalcemia, PTH normal o ligeramente elevada e hipocalciuria | |
| Hipercalcemia hipocalciúrica familiar (HHF) | AD | HPP neonatal severo: hipercalcemia grave y eventualmente hipercalciuria | ||
| HPP neonatal severo | AR | Mutaciones en los genes APS21 y GNA11 (que codifican proteínas implicadas en la señalización post-receptor) también se han relacionado con HHF | ||
| CGM2 | 6p24.2 | AD? | Protooncogén | Factor de transcripción implicado en el desarrollo de las glándulas paratiroides |
| AIP(proteína moduladora del receptor de aril hidrocarburos) | 11q13.2 | AD | Supresor tumoral | Su mutación en línea germinal se ha relacionado ampliamente con la aparición de adenomas hipofisarios (Familial Isolated Pituitary Adenoma [FIPA]). En 2013 se documentó mutación en línea germinal en dos casos de hiperparatiroidismo primario |
AD: autosómica dominante; AR: autosómica recesiva; TNE: tumores neuroendocrinos.
Los estudios de búsqueda sistemática de mutaciones en pacientes con HPP de riesgo han mostrado rendimiento variable. A nivel europeo destaca el estudio de Vierimaa et al.10, que en 29 pacientes finlandeses con datos sugestivos de HPP familiar (historia familiar, debut a edad <40años, enfermedad multiglandular, otros tumores sugestivos de síndrome neoplásico pluriglandular) analizó los genes MEN1, CDC73, CASR, AIP y CDKN1B, encontrando en un solo paciente una mutación en MEN1. En población americana destaca el estudio de Starker et al.11, que en 86 pacientes con HPP diagnosticado antes de los 45años y sin otros datos sindrómicos analizó los genes MEN1, CASR y CDC73, evidenciando en 8 de ellos mutaciones deletéreas (4MEN1, 3CASR y 1CDC73). Los autores concluyeron que el estudio genético en pacientes diagnosticados de HPP antes de los 45años es relevante y debe ser ofrecido a todos los pacientes como una rutina clínica. En población japonesa, el estudio de Mizusawa et al.12 analizó 13 familias con hiperparatiroidismo familiar con fenotipos variables, y encontraron 3 familias con mutaciones en CDC73 y una una familia con mutación en MEN1. Añaden como fenotipo de riesgo para mutaciones en CDC73 los adenomas paratiroideos quísticos. En 2020 se ha publicado el estudio más amplio hasta la fecha13, analizando en 121 sujetos británicos con perfil de riesgo un panel compuesto por los genes MEN1, CDC73, CASR, CDKN1A, CDKN1B, CDKN2B, CDKN2C, RET, GCM2, GNA11 y AP2S1, y que mostró presencia de variantes patogénicas en 19 pacientes (16%).
Nuestro grupo ha llevado a cabo un estudio en 40 sujetos de Sabadell con HPP y algún elemento de riesgo (edad ≤45años, historia familiar, histología de riesgo, tumor asociado, enfermedad multiglandular o HPP recidivado) que analiza un panel de genes compuesto por MEN1, RET, CDC73, CDKN1B, CDKN2B, CDKN2C, CASR, APS21, GNA11, AIP y GCM2 y ha identificado mutaciones en línea germinal —patogénicas o variantes de significado incierto— en 9 sujetos (22%)14.
Como conclusión, podemos decir que el estudio genético debe implementarse en la práctica cínica en nuestro medio en los pacientes con HPP que asocien algún elemento de sospecha de cuadro familiar, ya que en la actualidad disponemos de mayor evidencia sobre cómo hacer este cribado, y además el coste económico del análisis de estos paneles de genes se ha reducido de forma significativa.
