




The term pseudohypoparathyroidism (PHP) refers to a group of disorders characterized by resistance to the action of parathormone (PTH). This resistance appears in some cases as a result of GNAS gene alterations. It can cause hypocalcemia and hyperphosphatemia with high serum PTH levels in the presence of normal levels of 25-OH-Vitamin D, and a distinctive phenotype called “Albright hereditary osteodystrophy” (AHO) composed by short stature, round face, subcutaneous ossifications and brachydactyly. Given the clinical and radiological overlap between the classic PHP subtypes, and also between PHP and other phenotypically related skeletal disorders caused by alterations in other genes, a new classification proposes to incorporate all the patients affected by impairments in the same intracellular signaling pathway into a unifying clinical diagnose, named “Inactivating PTH/PTHrP signaling pathway disorders (iPPSD)” and split subtypes according only to molecular defects.1
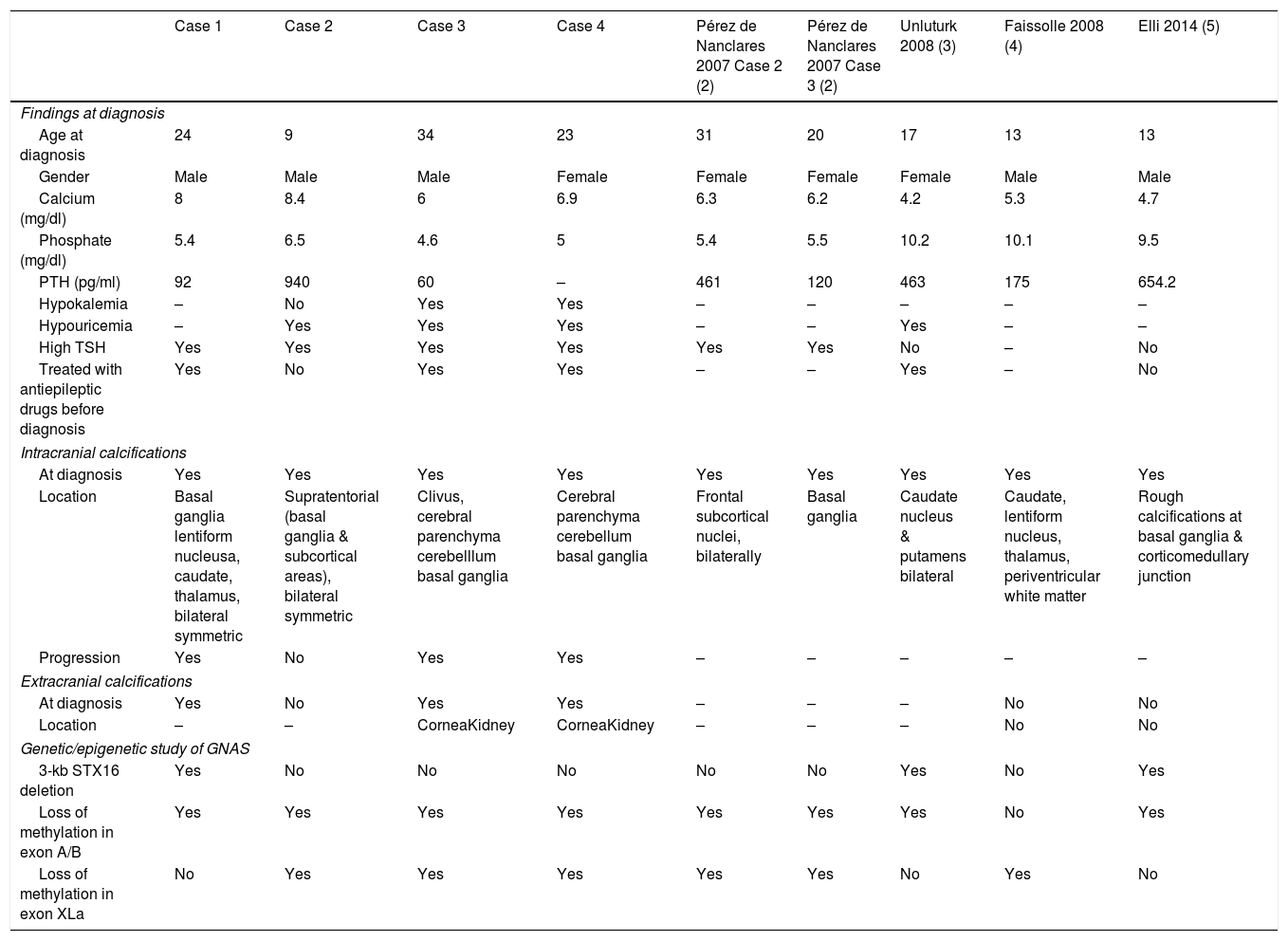
In the classic classification the presence of AHO was by definition absent in PHP1B, and the description of intracranial calcifications (IC) in patients diagnosed with PHP1A was not uncommon. However, up to now, there are only five reported cases of PHP1B, confirmed by genetic study, with IC: Two women, one 31-year-old and another 20-year-old,2 a 17-year old girl3 and two 13-year-old males.4,5 We describe here four unrelated patients diagnosed with PHP1B -and demonstrated GNAS epigenetic defects- that presented IC already at diagnosis. Table 1 summarizes the main clinical, biochemical and genetic findings of our four patients, together with the five other with PHP1B and IC previously described.3–5
Clinical, biochemical and genetic characteristics of the patients diagnosed with PHP1b and intracranial calcifications.
| Case 1 | Case 2 | Case 3 | Case 4 | Pérez de Nanclares 2007 Case 2 (2) | Pérez de Nanclares 2007 Case 3 (2) | Unluturk 2008 (3) | Faissolle 2008 (4) | Elli 2014 (5) | |
|---|---|---|---|---|---|---|---|---|---|
| Findings at diagnosis | |||||||||
| Age at diagnosis | 24 | 9 | 34 | 23 | 31 | 20 | 17 | 13 | 13 |
| Gender | Male | Male | Male | Female | Female | Female | Female | Male | Male |
| Calcium (mg/dl) | 8 | 8.4 | 6 | 6.9 | 6.3 | 6.2 | 4.2 | 5.3 | 4.7 |
| Phosphate (mg/dl) | 5.4 | 6.5 | 4.6 | 5 | 5.4 | 5.5 | 10.2 | 10.1 | 9.5 |
| PTH (pg/ml) | 92 | 940 | 60 | – | 461 | 120 | 463 | 175 | 654.2 |
| Hypokalemia | – | No | Yes | Yes | – | – | – | – | – |
| Hypouricemia | – | Yes | Yes | Yes | – | – | Yes | – | – |
| High TSH | Yes | Yes | Yes | Yes | Yes | Yes | No | – | No |
| Treated with antiepileptic drugs before diagnosis | Yes | No | Yes | Yes | – | – | Yes | – | No |
| Intracranial calcifications | |||||||||
| At diagnosis | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Location | Basal ganglia lentiform nucleusa, caudate, thalamus, bilateral symmetric | Supratentorial (basal ganglia & subcortical areas), bilateral symmetric | Clivus, cerebral parenchyma cerebelllum basal ganglia | Cerebral parenchyma cerebellum basal ganglia | Frontal subcortical nuclei, bilaterally | Basal ganglia | Caudate nucleus & putamens bilateral | Caudate, lentiform nucleus, thalamus, periventricular white matter | Rough calcifications at basal ganglia & corticomedullary junction |
| Progression | Yes | No | Yes | Yes | – | – | – | – | – |
| Extracranial calcifications | |||||||||
| At diagnosis | Yes | No | Yes | Yes | – | – | – | No | No |
| Location | – | – | CorneaKidney | CorneaKidney | – | – | – | No | No |
| Genetic/epigenetic study of GNAS | |||||||||
| 3-kb STX16 deletion | Yes | No | No | No | No | No | Yes | No | Yes |
| Loss of methylation in exon A/B | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No | Yes |
| Loss of methylation in exon XLa | No | Yes | Yes | Yes | Yes | Yes | No | Yes | No |
Case 1. A 39-year-old man was sent for hypocalcemia at the age of 24. He was already taking an antiepileptic drug—Carbamazepine—as recommended by his neurologist. A computerized axial tomography (CT) performed at PHP1B diagnosis in 2004 showed IC, and a brain MRI 4 years later confirmed their presence and progression. Case 2. A 38-year-old man with childhood obesity, and short stature presented at the age of 9 with mild hypocalcemia, hyperphosphatemia and elevated serum PTH levels. A brain CT scan performed at 22 years of age revealed IC, that persisted in another MRI done at 26 years of age. Case 3. A 60-year-old man had partial epileptic seizures that started at 23 years of age and were treated with Carbamazepine until PHP diagnosis, high blood pressure diagnosed at 31 years of age, bilateral cataracts revealed at 33, and short stature. At 34 years of age he presented symptomatic severe hypocalcemia associated with mild hyperphosphatemia and PTH levels in the upper normal range. At PHP diagnosis, signs suggestive of IC in the basal ganglia were observed. A cerebral CT scan performed 20 years later (at 54 years of age) indicated progression of the IC, as well as a sclerotic expansive bone lesion in the clivus. Currently, he presents moderate extrapyramidal symptoms that have required management by the neurologist. Case 4. A 65-year-old woman was diagnosed with PHP at the age of 23 based on hypocalcemia, hyperphosphatemia and the absence of response of cAMP to PTH. She had been previously diagnosed and treated for epilepsy with Diphenylhydantoin, due to the occurrence of cramps and carpopedal spasms, that started at the age of 17 years old, but persisted despite antiepileptic treatment. Her PHP1B diagnosis was ratified at 44 after carrying out a GNAS molecular genetic/epigenetic study. The presence of IC was confirmed at 23 years of age. At 60 years of age, a cerebral CT and an MRI were performed, describing progression of her IC. She has recently started with subtle extrapyramidal symptoms.
The early presence of IC in all cases, as well as the evidence of their progression in three of them (cases 1, 3 and 4) and associated evolving extrapyramidal manifestations in the oldest two (cases 3 and 4), highlights the importance of their early suspicion and detection, and provide new evidence to support the use of the new iPPSD classification for PHP disorders.1 The local and systemic metabolic mechanisms that underlie the development of symmetrical bilateral calcifications in the basal ganglia and cerebellum, have recently attracted close attention. It's been discovered that pericytes are able to initiate ectopic calcification in various tissues, and that mutations in the gene encoding PDGF-B (located at 22q13.1), impair pericyte function causing IC.6–8
Interestingly, treatment with antiepileptic drugs before PHP1B diagnosis was present in three of our patients, and in Unluturk's case.3 Misdiagnosis of epilepsy in PHP patients might be explained by two factors: the possibility of epileptiform activity in the EEG performed during the hypocalcemic convulsive crisis, and the absence of overt hypocalcemia if measured after the seizure. In fact, antiepileptic therapies before PHP diagnosis were also used in 2 siblings that presented with paroxysmal choreoathetosis finally found to have PHP1B.9 The knowledge of this typical misinterpretation of hypocalcemic PHP1B symptoms as epilepsy, could be useful as a hallmark to avoid delays in the correct diagnosis and treatments of the affected patients.
Additionally, persistent hypouricemia was evidenced in three of our patients, similarly to another two families with PHP1B previously reported,3,10 and a possible role for PTH resistance in the renal handling of uric acid has been suggested. Noteworthy, one of these reported patients with hypouricemia also harbored at PHP diagnosis IC of bilateral caudate nucleus and putamen.3 Besides, at diagnosis, our cases 3 and 4 also displayed hypokalemia.
Finally, despite the fact that the first international consensus statement on diagnosis and management of PHP and related disorders,11 in its recommendation 3.8 specifies that “for the evaluation of long-term consequences of hypocalcemia and hyperphosphatemia a brain CT is indicated only when neurological manifestations are present”, and that another publication from the same group,12 restate this approach, a recent review proposes the use of systematic screening of IC with head CT in all apparently asymptomatic patients with PHP of long duration.6 Our findings support this proposal, and suggest that IC screening of patients with PHP1B could also be useful at diagnosis, even in the absence of evident neurological impairment.
FundingThis research has received no funding.
Conflict of interestThe authors declare that they have no conflict of interest.
