




El linfoma primario tiroideo (LPT) es una entidad rara que se define por la afectación única de la glándula tiroides y de los ganglios linfáticos locorregionales, debiéndose descartar la afectación en otra ubicación en el momento del diagnóstico1. Representa menos del 2-5% de las neoplasias de tiroides y menos del 2,5% de los linfomas extranodales2. Presenta una mayor prevalencia en mujeres con una relación 4:1 y se desarrolla en la mayoría de los casos entre los 60 y 75 años, con una edad media de 67 años3. La forma de presentación clínica más habitual es una masa de crecimiento rápido, que puede ser dolorosa, y que ocasiona síntomas por compresión (disnea, disfagia, afonía, estridor y tos). El 10-20% de los LPT desarrollan un cuadro sistémico asociado (fiebre, sudoración nocturna y pérdida de peso)4,5. Lo habitual es que los sujetos se encuentren eutiroideos en el momento del diagnóstico aunque un 10% pueden presentar un hipotiroidismo primario.
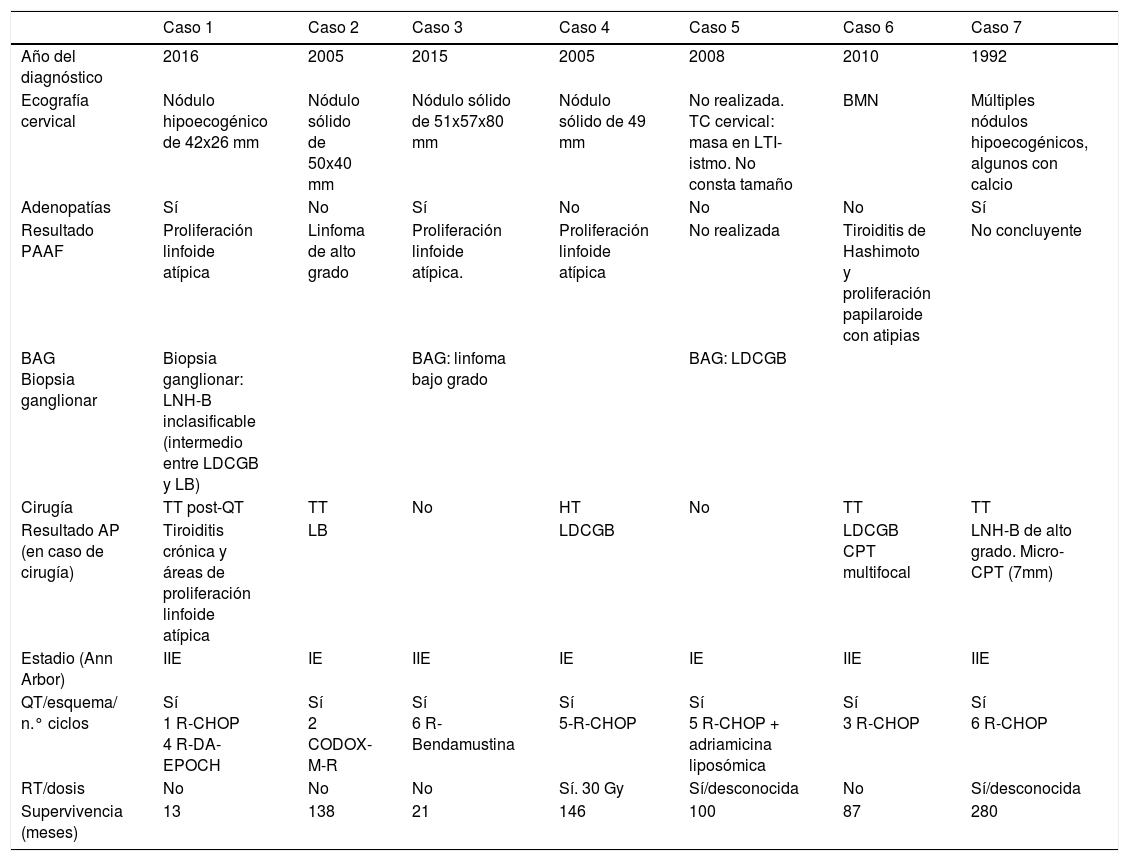
El objetivo de este trabajo es presentar nuestra experiencia en el manejo de los LPT en 3 hospitales de Castilla-La Mancha. Para ello se seleccionaron aquellos pacientes con diagnóstico histológico de LPT o con ese diagnóstico codificado en el informe de alta hospitalaria desde 1990 hasta la actualidad, y se realizó un análisis retrospectivo de las historias clínicas. Siete pacientes, todas mujeres con una edad media de 59 años, cumplían estas características. En todos los casos el LPT se presentó como un nódulo tiroideo no doloroso asociado a sintomatología compresiva de rápido crecimiento (1-12 semanas de evolución). En 6 casos (85,7%), las pacientes presentaban una tiroiditis crónica autoinmune asociada y en 4 casos (57%) un hipotiroidismo primario. Los resultados de las pruebas de imagen se muestran en la tabla 1. En 6 casos (85,7%) se realizó una punción aspiración con aguja fina (PAAF) guiada por ecografía; el resultado citológico se muestra en la tabla 1. En 2 pacientes, en uno de ellos tras PAAF, se realizó biopsia con aguja gruesa (BAG). Se realizó tratamiento quirúrgico en el 71% de los casos: tiroidectomía total (TT) en 4 pacientes y hemitiroidectomía en un caso. En todos los casos se administró quimioterapia (QT) sistémica; los esquemas de tratamiento se resumen en la tabla 1. Tres pacientes (42,8%) recibieron también radioterapia (RT). Por último, en ningún caso se ha producido muerte o recidiva del LPT.
Características de los casos de linfoma primario tiroideo
| Caso 1 | Caso 2 | Caso 3 | Caso 4 | Caso 5 | Caso 6 | Caso 7 | |
|---|---|---|---|---|---|---|---|
| Año del diagnóstico | 2016 | 2005 | 2015 | 2005 | 2008 | 2010 | 1992 |
| Ecografía cervical | Nódulo hipoecogénico de 42x26 mm | Nódulo sólido de 50x40 mm | Nódulo sólido de 51x57x80 mm | Nódulo sólido de 49 mm | No realizada. TC cervical: masa en LTI-istmo. No consta tamaño | BMN | Múltiples nódulos hipoecogénicos, algunos con calcio |
| Adenopatías | Sí | No | Sí | No | No | No | Sí |
| Resultado PAAF | Proliferación linfoide atípica | Linfoma de alto grado | Proliferación linfoide atípica. | Proliferación linfoide atípica | No realizada | Tiroiditis de Hashimoto y proliferación papilaroide con atipias | No concluyente |
| BAG Biopsia ganglionar | Biopsia ganglionar: LNH-B inclasificable (intermedio entre LDCGB y LB) | BAG: linfoma bajo grado | BAG: LDCGB | ||||
| Cirugía | TT post-QT | TT | No | HT | No | TT | TT |
| Resultado AP (en caso de cirugía) | Tiroiditis crónica y áreas de proliferación linfoide atípica | LB | LDCGB | LDCGB CPT multifocal | LNH-B de alto grado. Micro-CPT (7mm) | ||
| Estadio (Ann Arbor) | IIE | IE | IIE | IE | IE | IIE | IIE |
| QT/esquema/ n.° ciclos | Sí 1 R-CHOP 4 R-DA-EPOCH | Sí 2 CODOX-M-R | Sí 6 R-Bendamustina | Sí 5-R-CHOP | Sí 5 R-CHOP + adriamicina liposómica | Sí 3 R-CHOP | Sí 6 R-CHOP |
| RT/dosis | No | No | No | Sí. 30 Gy | Sí/desconocida | No | Sí/desconocida |
| Supervivencia (meses) | 13 | 138 | 21 | 146 | 100 | 87 | 280 |
AP: anatomía patológica; BAG: biopsia con aguja gruesa; BMN: bocio multinodular; CODOX-M-R: ciclofosfamida, doxorrubicina, prednisona, vincristina, metotrexate, rituximab; CPT: carcinoma papilar de tiroides; HT: hemitiroidectomía; LB: linfoma de Burkitt; LDCGB: linfoma difuso de células grandes B; LNH-B: linfoma no Hodgkin B; LTI: lóbulo tiroideo izquierdo; PAAF: punción aspiración con aguja fina; QT: quimioterapia; R-CHOP: rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona; R-DA-EPOCH: rituximab, etopósido, prednisona, vincristina, ciclofosfamida y doxorrubicina; RT radioterapia; TC tomografía computarizada; TT: tiroidectomía total.
Los LPT son neoplasias infrecuentes. La mayoría de los LPT son linfomas no Hodgkin (LNH). El 50-80% son linfomas difusos de células grandes B (LDCGB) y el 20-30% son de tejido linfoide asociado a mucosas (MALT), mientras que otros subtipos histológicos como el folicular, linfocítico de célula pequeña, Burkitt (LB), Hodgkin o linfoma T son extremadamente infrecuentes6. En nuestra serie, una paciente presenta un LB y en dos casos coexistían además carcinomas papilares de tiroides (CPT). Aunque el CPT representa el 85% de los cánceres del epitelio folicular del tiroides, la asociación de LPT y CPT es excepcional y hay pocos casos descritos en la literatura7,8. El riesgo de presentar un LPT se multiplica por 80 en presencia de una tiroiditis crónica autoinmune, aunque la evolución de esta entidad a linfoma es infrecuente1. Por otra parte, la relación de la tiroiditis crónica autoinmune con el CPT sigue siendo objeto de controversia aunque su coexistencia representa una realidad clínica cuyo significado aún desconocemos9. El diagnóstico de certeza de los LPT a menudo requiere una biopsia quirúrgica, ya que la mayoría de las exploraciones citológicas presentan una baja sensibilidad10. El papel de la PAAF en el diagnóstico del LPT es limitado por la dificultad en realizar el diagnóstico diferencial entre linfoma y la infiltración linfocitaria tiroidea. Sin embargo, la sensibilidad de la PAAF ha aumentado considerablemente al introducir otras técnicas como la citometría de flujo, estudios inmunohistoquímicos o técnicas moleculares. En nuestra serie, únicamente en un caso se obtuvo el diagnóstico de certeza tras PAAF (caso 2); se trataba de una paciente con un LB, que supone una variante más agresiva. En el caso 1 se realizó citometría de flujo en el material obtenido de la PAAF y esta técnica ayudó a completar el diagnóstico de LNH B, aunque finalmente también se realizó una biopsia ganglionar para obtener el diagnóstico anatomopatológico definitivo. En los casos 3 y 5 se realizaron técnicas de biología molecular en la muestra obtenida por BAG, confirmando así el diagnóstico anatomopatológico sin requerir cirugía. La estadificación se realiza según la clasificación de Ann Arbor: IE (enfermedad limitada al tiroides), IIE (afectación de tiroides y ganglios locorregionales), IIIE (afectación de ganglios a ambos lados del diafragma) e IVE (afectación difusa). El 90% de los LPT se diagnostican en estadios tempranos de la enfermedad11, tal y como sucede en nuestra serie.
Tradicionalmente, el tratamiento de los LPT era la cirugía. En nuestra serie en cinco casos se optó por tratamiento quirúrgico. En todos los casos, el tratamiento quirúrgico fue previo a la administración de QT sistémica salvo en el caso 1, en el que se practicó una TT tras el tratamiento con QT sistémica al sospechar persistencia de enfermedad por captación tiroidea en la tomografía por emisión de positrones (PET-TC) con 18-fluorodesoxiglucosa (18FDG), aunque finalmente el resultado anatomopatológico fue de tiroiditis crónica, descartando así la persistencia de enfermedad tras el tratamiento recibido. En el resto de casos la cirugía tuvo una indicación terapéutica (una paciente tenía un LB, que supone una variante más agresiva, en otros casos el estudio citológico no fue concluyente y en el último caso el resultado de la PAAF hacía sospechar un CPT, por lo que se optó por realizar una TT). En la actualidad el tratamiento de elección es la QT sistémica según el esquema CHOP (añadiendo rituximab en los LNH B) con o sin RT asociada12, aunque no existen estudios prospectivos aleatorizados debido que se trata de una patología rara. En nuestro caso, en dos situaciones se optó por otro esquema terapéutico de QT más intensivo ya que las pacientes presentaban linfomas con un pronóstico más desfavorable. El pronóstico de los LPT depende del tipo histológico y del estadio de la enfermedad. Los LNH tipo MALT presentan un mejor pronóstico debido a su comportamiento más indolente. Por otro lado, la tasa de supervivencia a los 5 años es del 80% para el estadio IE y del 50 y 35% para los estadio IIE y III-IVE respectivamente1,13. Cabe destacar que en nuestra serie todos los pacientes han alcanzado remisión de la enfermedad con una supervivencia del 100%.
En conclusión, aunque el LPT se trata de una entidad muy infrecuente, debe sospecharse en mujeres con un crecimiento tiroideo rápido asociado a sintomatología compresiva. La mayoría son LNH-B que se diagnostican en estadios precoces de la enfermedad y en estos casos la tasa de supervivencia es elevada. El tratamiento de elección es la QT sistémica mientras que la cirugía tiene un papel fundamentalmente diagnóstico.
