






Serum phosphorus levels range from 2.5 and 4.5mg/dl (0.81–1.45mmol/l) in adults, with higher levels in childhood, adolescence, and pregnancy. Intracellular phosphate is involved in intermediary metabolism and other essential cell functions, while extracellular phosphate is essential for bone matrix mineralization. Plasma phosphorus levels are maintained within a narrow range by regulation of intestinal absorption, redistribution, and renal tubular absorption of the mineral. Hypophosphatemia and hyperphosphatemia are common clinical situations, although changes are most often mild and oligosymptomatic. However, acute and severe conditions that require specific treatment may occur. In this document, members of the Mineral and Bone Metabolism Working Group of the Spanish Society of Endocrinology and Nutrition review phosphate disorders and provide algorithms for adequate clinical management of hypophosphatemia and hyperphosphatemia.
La concentración sérica de fósforo oscila entre 2,5 y 4,5mg/dl (0,81-1,45mmol/l) en adultos, con niveles más altos en la infancia, la adolescencia y durante la gestación. El fosfato intracelular está implicado en el metabolismo intermediario y otras funciones celulares esenciales, mientras que el extracelular es fundamental para la mineralización de la matriz ósea. La fosforemia se mantiene en un estrecho rango mediante la regulación de la absorción intestinal, la redistribución y la reabsorción tubular renal de fósforo. La hipofosfatemia y la hiperfosfatemia son situaciones clínicas frecuentes, aunque, en la mayoría de las ocasiones, se trata de alteraciones leves y poco sintomáticas. Sin embargo, pueden presentarse cuadros agudos y severos que requieren tratamiento específico. En este documento elaborado por miembros del Grupo de Trabajo de Metabolismo Mineral y Óseo de la Sociedad Española de Endocrinología y Nutrición se revisan los trastornos del fosfato y se proporcionan algoritmos de manejo clínico de la hipofosfatemia y la hiperfosfatemia.
Phosphorus accounts for 1% of total body weight. The body reserves of organic phosphorus are essentially located in bone (85%) as hydroxyapatite crystals. Most other phosphates are intracellular (14%), and less than 1% of total phosphorus is found in extracellular fluid. Circulating phosphate is present as a univalent or divalent hydrogenated species (the univalent form being four times more common). Normal serum phosphorus levels range from 2.5–4.5mg/dl (0.81–1.45mmol/l) in adults, with higher levels in childhood, adolescence, and pregnancy. Intracellular phosphate is involved in intermediary metabolism and other essential cell functions, while extracellular phosphate is essential for the mineralization of the bone matrix.1,2 Serum phosphorus levels are kept within a narrow range through interrelated changes in intestinal absorption, redistribution between compartments (intracellular, extracellular and bone), and renal tubular reabsorption.1–4
The most common foods in the diet (dairy products, legumes, cereals, meat, fish, soft drinks, etc.) are rich in phosphorus (those of animal origin being more bioavailable), and the dietary supply and absorbed fraction are usually higher than the daily requirements (580–1055mg/day). Between 60 and 70% of dietary phosphate is absorbed in the small intestine (especially in the jejunum) through sodium-dependent and -independent pathways that may involve vitamin D.1,3
The bone phosphate reservoir mobilizes in a way similar to the calcium reservoir, though in addition to parathyroid hormone (PTH) and vitamin D, such mobilization is mediated by other hormones such as fibroblast growth factor 23 (FGF23).1–4
With regard to renal processing, under normal conditions most filtered phosphate is reabsorbed in the proximal tubule, while 10–20% is excreted in urine. Reabsorption in the proximal tubule is mediated by three different transport proteins dependent upon concomitant sodium transport.1–4
While intestinal absorption and bone resorption are relevant, renal tubular reabsorption is the determinant mechanism underlying blood phosphate levels. Renal phosphate reabsorption depends on a number of factors, particularly dietary phosphate intake, blood phosphate concentration, and the activity of the hormones implicated in its control: PTH, FGF23, other phosphatonins and vitamin D.
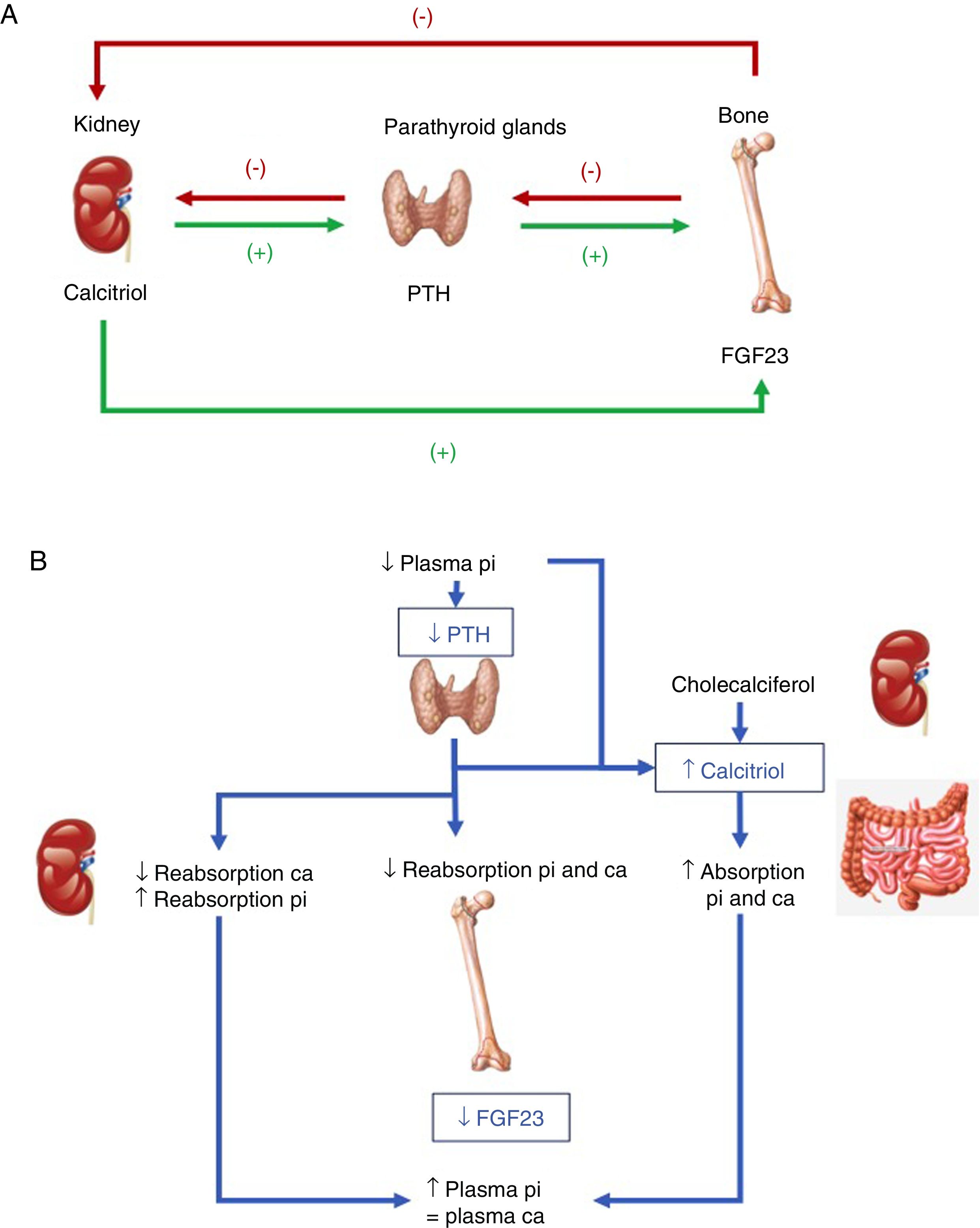
Hormones involved (Fig. 1):
- •
PTH: its main stimulus is hypocalcemia at parathyroid calcium sensing receptor level, causing the renal elimination of phosphorus. Accordingly, after inducing greater osteoclastic action in bone, which releases calcium and phosphorus from bone hydroxyapatite, there is a rise in blood calcium levels, but not phosphate levels. In addition, PTH activates 1-α-hydroxylase for the synthesis of active vitamin D or calcitriol.
- •
Vitamin D: to generate its active form, the hormonal precursor produced in the skin (vitamin D3, 80%) or obtained through the diet (vitamin D2 or D3, 20%) is activated via two consecutive hydroxylation steps: in the liver, at position 25 (scantly regulated), and in the kidneys, at position 1-α. The latter step is stimulated by hypocalcemia and hypophosphatemia, and by the regulatory hormones PTH, calcitonin, growth hormone (GH) and insulin growth factor 1, and is inhibited by FGF23, its product, calcitriol, and by hyperphosphatemia. Vitamin D also increases calcium and phosphorus absorption (the latter through unknown mechanisms), and may modulate both bone resorption activating and inhibiting actions, possibly through an increase in the receptor activator of nuclear factor kappa-B ligand (RANKL). In the kidney, it increases tubular calcium reabsorption and stimulates the production of FGF23.
- •
FGF23: FGF23-Klotho is a peptide hormone produced by osteocytes, osteoblasts and mesenchymal cells in response to high levels of phosphorus and calcitriol. The Klotho cofactor is required to conform its receptor. FGF23 causes phosphaturia by inhibiting phosphorus tubular reabsorption through inhibition of the expression of the corresponding transporters, and also inhibits 1-α-hydroxylase and activates 24-hydroxylase, reducing calcitriol levels and phosphate (and calcium) absorption.
 Tissues and hormones involved in the regulation of phosphate metabolism. (B) Integrated response to correct hypocalcemia. Adapted from Fukumoto.4")
(A) Tissues and hormones involved in the regulation of phosphate metabolism. (B) Integrated response to correct hypocalcemia. Adapted from Fukumoto.4
As regards the integration of hormonal action with dietary intake, both low phosphate intake and hypophosphatemia increase renal phosphate reabsorption. Unlike in the case of calcium, no phosphate-sensing receptor has been characterized in mammals, though both PTH and FGF23 – the main phosphaturic hormones – act by increasing phosphate excretion. The mechanism by which high dietary phosphate intake or hyperphosphatemia promotes the release of FGF23 and active D hormone is not known.2–4
Phosphate metabolism is finely regulated from the parathyroid glands, kidneys and bone, though the kidneys play a key role in modulating phosphate levels, and FGF23 emerges as the main phosphaturic hormone. A deficiency of FGF23 causes hyperphosphatemia, excess calcitriol and soft tissue calcifications, while its excess causes hypophosphatemia, hypofunction of the vitamin D system, and altered growth.1–4
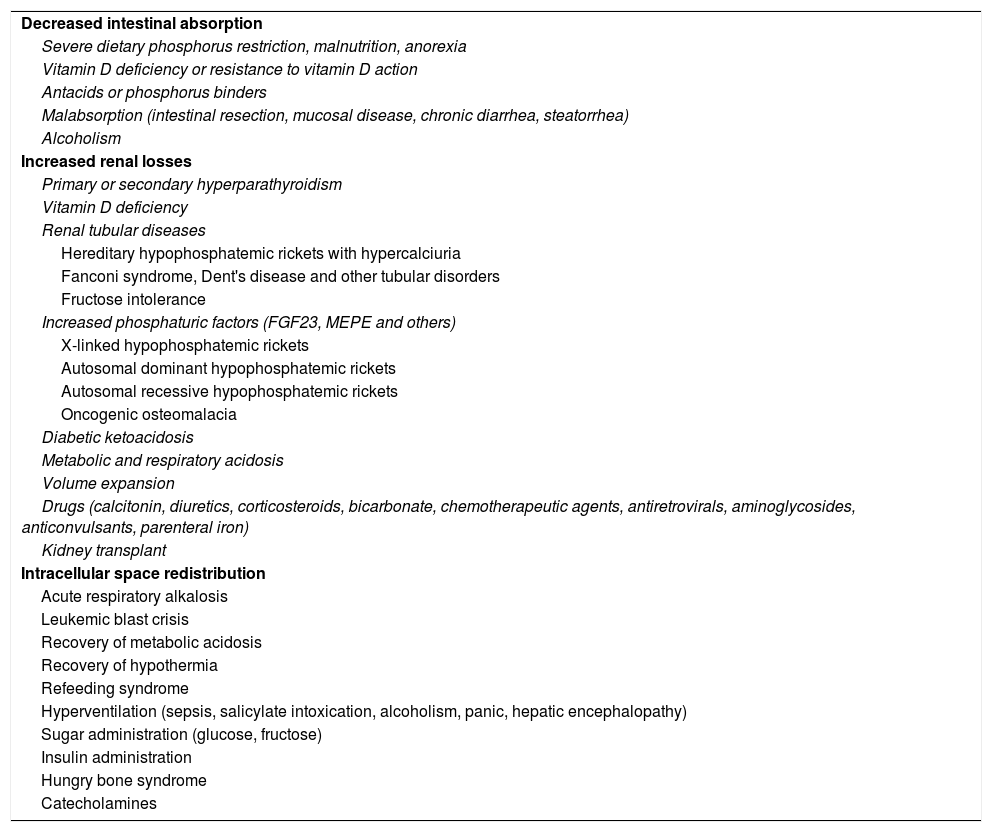
The etiology of hypophosphatemiaHypophosphatemia is defined as a serum phosphorus concentration <2.5mg/dl, and the condition may be mild (2.0–2.5mg/dl), moderate (1.0–2.0mg/dl) or severe (<1.0mg/dl). The main causes of hypophosphatemia are classified according to the underlying physiopathological mechanism4–10 (Table 1). In turn, hypophosphatemia can be classified as either acute or chronic8,11 (Table 2). Acute hypophosphatemia is usually due to phosphorus redistribution phenomena, while chronic hypophosphatemia is attributable to altered tubular reabsorption.
Mechanisms and etiopathogenic classification of hypophosphatemia.
| Decreased intestinal absorption |
| Severe dietary phosphorus restriction, malnutrition, anorexia |
| Vitamin D deficiency or resistance to vitamin D action |
| Antacids or phosphorus binders |
| Malabsorption (intestinal resection, mucosal disease, chronic diarrhea, steatorrhea) |
| Alcoholism |
| Increased renal losses |
| Primary or secondary hyperparathyroidism |
| Vitamin D deficiency |
| Renal tubular diseases |
| Hereditary hypophosphatemic rickets with hypercalciuria |
| Fanconi syndrome, Dent's disease and other tubular disorders |
| Fructose intolerance |
| Increased phosphaturic factors (FGF23, MEPE and others) |
| X-linked hypophosphatemic rickets |
| Autosomal dominant hypophosphatemic rickets |
| Autosomal recessive hypophosphatemic rickets |
| Oncogenic osteomalacia |
| Diabetic ketoacidosis |
| Metabolic and respiratory acidosis |
| Volume expansion |
| Drugs (calcitonin, diuretics, corticosteroids, bicarbonate, chemotherapeutic agents, antiretrovirals, aminoglycosides, anticonvulsants, parenteral iron) |
| Kidney transplant |
| Intracellular space redistribution |
| Acute respiratory alkalosis |
| Leukemic blast crisis |
| Recovery of metabolic acidosis |
| Recovery of hypothermia |
| Refeeding syndrome |
| Hyperventilation (sepsis, salicylate intoxication, alcoholism, panic, hepatic encephalopathy) |
| Sugar administration (glucose, fructose) |
| Insulin administration |
| Hungry bone syndrome |
| Catecholamines |
FGF23: fibroblast growth factor 23; MEPE: matrix extracellular phosphoglycoprotein.
Causes of hypophosphatemia.
| Acute hypophosphatemia |
| Increased renal losses |
| Following kidney transplant (first weeks) |
| Acute volume expansion |
| Intracellular space redistribution |
| Following bone marrow transplant |
| Acute leukemia and lymphoma |
| Correction of diabetic ketoacidosis |
| Osmotic diuresis |
| Refeeding syndrome |
| Acute respiratory alkalosis |
| Hungry bone syndrome |
| Miscellaneous |
| Severe sepsis |
| Major burns |
| Inadequate dialysis |
| Acute paracetamol overdose |
| Salicylate intoxication |
| Hypothermia |
| Chronic hypophosphatemia |
| Deficient supply |
| Inadequate parenteral nutrition |
| Nutritional deficiencies |
| Decreased intestinal absorption |
| Vitamin D deficiency |
| Vitamin D-dependent rickets |
| Chronic antacid therapy |
| Malabsorptive syndromes |
| Chronic kidney disease |
| Chronic alcoholism |
| Increased renal losses due to primary kidney disease |
| Fanconi syndrome |
| Dent's disease |
| Toxic agents (ifosfamide, platinum salts, diuretics, glucocorticoids, antiretrovirals, acetazolamide, aminoglycosides) |
| Primary or secondary hyperparathyroidism |
| Increased serum FGF23 levels |
| Hypophosphatemic rickets |
| Tumor-induced osteomalacia |
| Fibrous dysplasia |
| McCune-Albright syndrome |
| Toxic agents (parenteral iron) |
| Intracellular space redistribution |
| Treatment with erythropoiesis-stimulating agents in patients with cirrhosis |
FGF23: fibroblast growth factor 23.
Adapted from Bacchetta and Salusky.11
From the physiopathological perspective, three causes of hypophosphatemia may be established:
- a.
Diminished intestinal absorption of phosphorus: this situation is exceptional, due to the widespread abundance of phosphate in food.
- b.
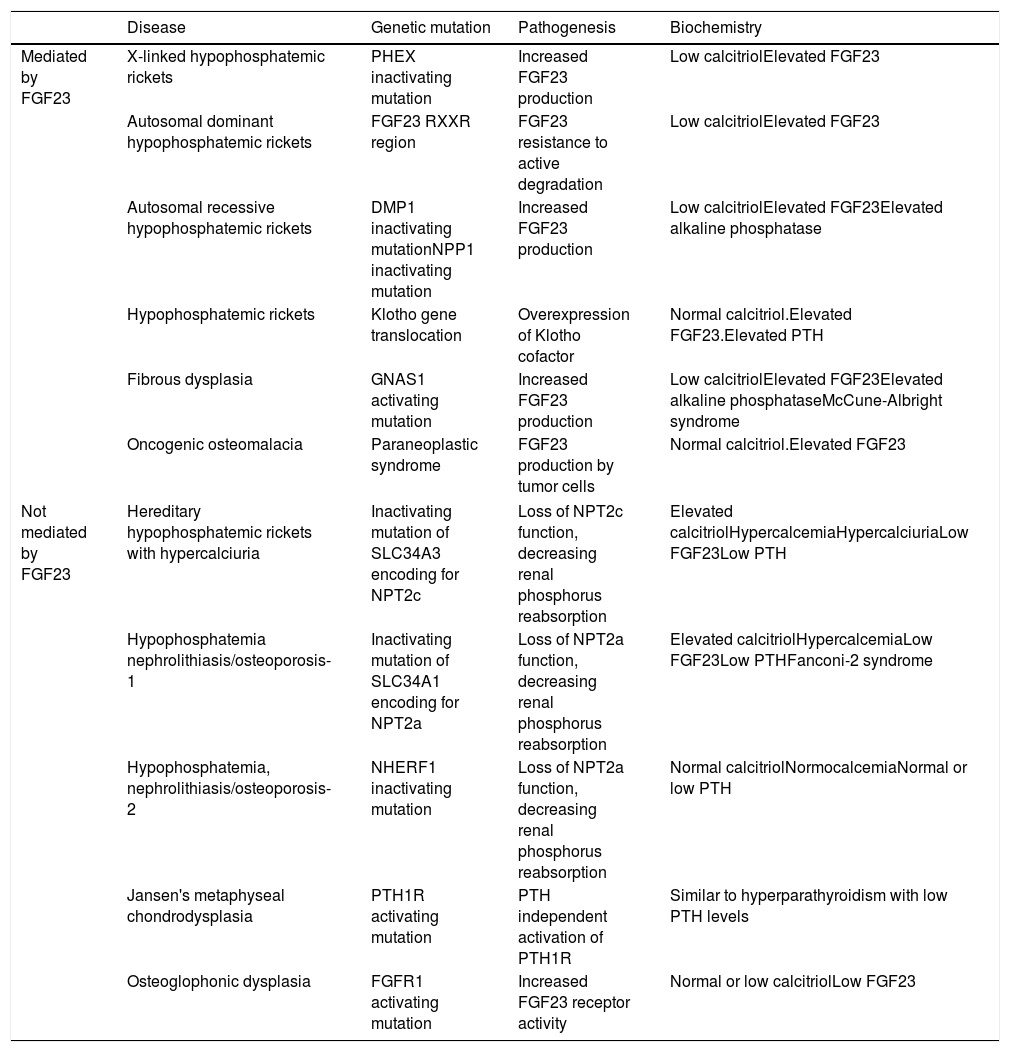
Increased renal phosphorus losses: this is the most common cause of hypophosphatemia. The condition may or may not be mediated by FGF23 (Table 3), and may be of genetic, neoplastic or acquired origin.12–14
Table 3.Causes of hypophosphatemia due to increased renal phosphate excretion.
Disease Genetic mutation Pathogenesis Biochemistry Mediated by FGF23 X-linked hypophosphatemic rickets PHEX inactivating mutation Increased FGF23 production Low calcitriolElevated FGF23 Autosomal dominant hypophosphatemic rickets FGF23 RXXR region FGF23 resistance to active degradation Low calcitriolElevated FGF23 Autosomal recessive hypophosphatemic rickets DMP1 inactivating mutationNPP1 inactivating mutation Increased FGF23 production Low calcitriolElevated FGF23Elevated alkaline phosphatase Hypophosphatemic rickets Klotho gene translocation Overexpression of Klotho cofactor Normal calcitriol.Elevated FGF23.Elevated PTH Fibrous dysplasia GNAS1 activating mutation Increased FGF23 production Low calcitriolElevated FGF23Elevated alkaline phosphataseMcCune-Albright syndrome Oncogenic osteomalacia Paraneoplastic syndrome FGF23 production by tumor cells Normal calcitriol.Elevated FGF23 Not mediated by FGF23 Hereditary hypophosphatemic rickets with hypercalciuria Inactivating mutation of SLC34A3 encoding for NPT2c Loss of NPT2c function, decreasing renal phosphorus reabsorption Elevated calcitriolHypercalcemiaHypercalciuriaLow FGF23Low PTH Hypophosphatemia nephrolithiasis/osteoporosis-1 Inactivating mutation of SLC34A1 encoding for NPT2a Loss of NPT2a function, decreasing renal phosphorus reabsorption Elevated calcitriolHypercalcemiaLow FGF23Low PTHFanconi-2 syndrome Hypophosphatemia, nephrolithiasis/osteoporosis-2 NHERF1 inactivating mutation Loss of NPT2a function, decreasing renal phosphorus reabsorption Normal calcitriolNormocalcemiaNormal or low PTH Jansen's metaphyseal chondrodysplasia PTH1R activating mutation PTH independent activation of PTH1R Similar to hyperparathyroidism with low PTH levels Osteoglophonic dysplasia FGFR1 activating mutation Increased FGF23 receptor activity Normal or low calcitriolLow FGF23 DMP1: dentin matrix acidic phosphoprotein 1; FGF23: fibroblast growth factor 23; FGFR1: fibroblast growth factor receptor 1; NPP1: nucleotide pyrophosphatase/phosphodiesterase 1; NPT2c: sodium-dependent phosphate transporter type 2c; NPT2a: sodium-dependent phosphate transporter type 2a; PTH: parathyroid hormone; PTH1R: parathyroid hormone receptor 1.
Other causes are treatment with diuretics such as acetazolamide, thiazides, loop diuretics and mannitol, as well as hyper- and normocalcemic hyperparathyroidism and secondary hyperparathyroidism associated with normal renal function with hypocalcemia.
- c.
Phosphorus displacement from the extracellular to the intracellular compartment: different situations or treatment can result in the stimulation of glycolysis, with increased cellular phosphate uptake and/or phosphate consumption secondary to increased anabolism.
The signs and symptoms of hypophosphatemia appear when the latter is accompanied by the depletion of intracellular phosphate, but not when hypophosphatemia is due only to phosphate displacement toward the cell. Thus, severe acute hypophosphatemia is seen mainly in hospitalized patients presenting severe medical and surgical conditions, with acute deficiency and pre-existing phosphate depletion. Acute hypophosphatemia without prior chronic phosphate depletion is usually not symptomatic.15
1. The effects upon mineral metabolism:
- –
At the renal level: inhibition of the tubular reabsorption of calcium and magnesium, causing hypercalciuria.16
- –
At bone level: increased bone resorption, with an increase in serum calcium levels that contributes to hypercalciuria. Rickets and osteomalacia may develop over the long term.17
2. The effects upon other systems due to deficient adenosine triphosphate and 2–3 diphosphoglycerate in erythrocytes:
The decrease in red blood cell 2–3 diphosphoglycerate levels increases hemoglobin affinity for oxygen and reduces oxygen release at the tissue level. The decrease in adenosine triphosphate in turn results in changes in cell functions that require this molecule in order to obtain energy. These alterations result in symptoms at different levels:
- –
The central nervous system (CNS): from mild irritability and paresthesia to more serious manifestations in the form of metabolic encephalopathy, delirium, generalized seizures and coma.18 This condition may also contribute to the development of central pontine myelinolysis.19
- –
The cardiopulmonary system: impaired myocardial contractility, especially in patients with severe hypophosphatemia (<1mg/dl). This condition has also been associated with ventricular arrhythmias and an increased need for vasoactive drugs in heart surgery.20 It also affects diaphragmatic contractility and thus lung function.21
- –
Skeletal and smooth muscle: proximal myopathy, dysphagia and ileus. Episodes of rhabdomyolysis have also been reported in refeeding syndrome and in alcoholics.22
- –
Hematological manifestations: hemolysis and impaired leukocyte function (reduction of phagocytosis and granulocyte chemotaxis). These situations are rare and occur at very low phosphorus levels (<0.5mg/dl). Platelet alterations with defective clot retraction and thrombocytopenia may also develop.15
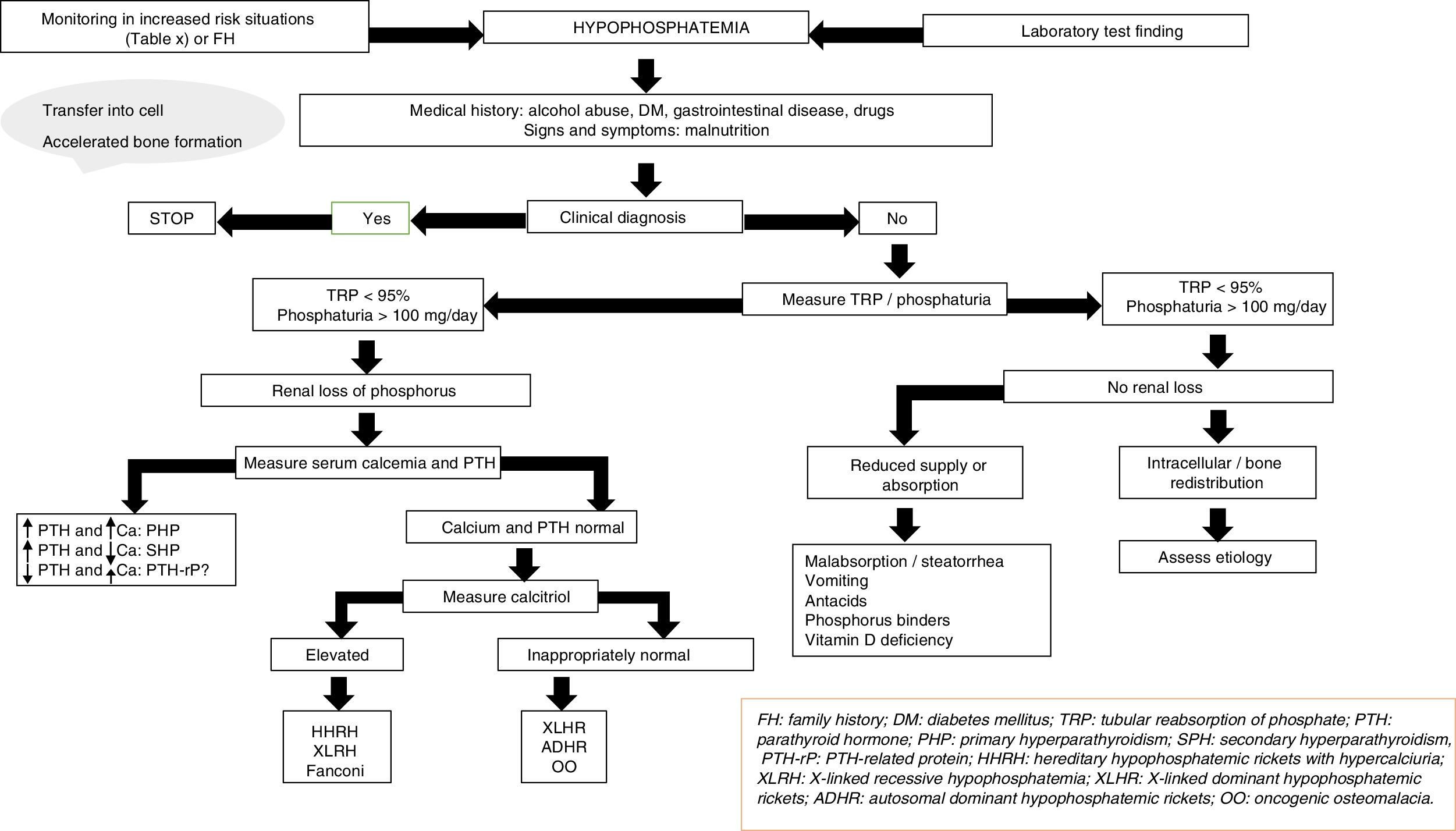
The etiology of hypophosphatemia is often established from a correct case history. In all other cases, and in the absence of pseudo-hypophosphatemia due to paraproteins,23 urine phosphate excretion should be measured in 24-h urine; alternatively, the tubular reabsorption of phosphorus can be calculated in an isolated urine sample. The physiological response at the renal level to a decrease in serum phosphate consists of an increase in phosphate reabsorption, which virtually suppresses phosphaturia.
In this regard, the presence of phosphate levels below 100mg daily in 24-h urine, or phosphorus tubular reabsorption >95%, suggests an extrarenal cause, including in particular internal phosphate redistribution or a decrease in phosphate intestinal absorption.15 Intracellular redistribution is more common in patients receiving glucose or insulin infusions, as occurs in refeeding syndrome or in the treatment of acute hyperglycemia in diabetes mellitus. It may also occur in patients with hyperventilation and acute respiratory alkalosis. At the intestinal level, different mechanisms may result in decreased phosphate absorption, increased transit and secretions in chronic diarrhea, the formation of insoluble salts in treatment with calcium, magnesium, or aluminum supplements, and the inhibition of intestinal transport in concomitant niacin therapy.15
Moreover, phosphaturia ≥100mg daily or a percentage tubular reabsorption of phosphorus <95% is indicative of renal phosphate loss and suggests excess phosphaturic peptides or hormones (PTH, FGF23, Klotho), altered vitamin D metabolism or, to a lesser extent, primary tubular defects. Both primary and secondary hyperparathyroidism may be associated with hypophosphatemia.15 The measurement of serum and urinary calcium, kidney function, magnesium levels, vitamin D, and drug use, etc. will help establish the differential diagnosis between the two conditions. The determination of PTH-related protein (PTH-rp) should be made under conditions of hypercalcemia with low intact PTH levels.
In the absence of hyperparathyroidism and altered calcium levels, the presence of genetic forms of hypophosphatemic rickets or of oncogenic osteomalacia (OO), a paraneoplastic syndrome, should be considered.24,25 Patient age at onset, the clinical profile, calcitriol and FGF23 levels, and a request for genetic tests will help establish a differential diagnosis among the different conditions (Fig. 2).

X-linked hypophosphatemic rickets (XLHR), autosomal dominant hypophosphatemic rickets (ADHR) and OO are associated with decreased or inappropriately normal calcitriol levels and, in general, with elevated FGF23 levels.25 High levels of this hormone imply a decrease in renal phosphate reabsorption and in the compensatory synthesis of 1,25-dihydroxyvitamin D. In children, genetic analysis of the PHEX gene contributes to the diagnosis of XLHR, though the absence of known mutations does not rule out the disease, since they have been shown to be present in only 50–70% of all patients.26 A differential diagnosis should be established with autosomal recessive hypophosphatemic rickets, which is an extremely rare condition associated with findings similar to XLHR, and which usually manifests in late childhood. Analysis of the genes responsible for the synthesis of proteins related to FGF23 degradation serves to establish the diagnosis of the three subtypes known to date.27,28
In adults, the finding of previous normal blood phosphate levels demonstrates the presence of OO, though sometimes patients with ADHR may also exhibit normal levels.25 Oncogenic osteomalacia is caused by mesenchymal tumors with ectopic secretion of FGF23 and other phosphatonins. Locating the culprit tumor is often a diagnostic challenge in which different radiological and nuclear techniques have been used. A stepwise diagnostic approach has recently been proposed, involving positron emission tomography-computed tomography (PET-CT) with somatostatin analog peptides such as DOTA0-Tyr3-octreotate labeled with gallium 68, systemic venous sampling of FGF23, and 3T magnetic resonance imaging (MRI).29 Until this diagnostic approach becomes a reality in standard clinical practice, initial assessment should remain based on the usual techniques (Octreoscan, PET-CT with fluorodeoxyglucose, computed tomography or MRI). Although ADHR is very uncommon, the absence of OO requires a genetic study of the condition. Different mutations of the FGF23 gene have been associated with FGF23 resistance to proteolytic degradation. However, FGF23 levels are not consistently elevated. This has been attributed to the presence of quiescent periods in which both phosphorus and FGF23 levels are normal in this group of patients.30 In turn, more recent studies have associated iron deficiency with high FGF23 levels in patients with ADHR.31
Lastly, a number of tubular defects have been associated with increased renal phosphate loss.15,25 In addition to hypophosphatemia, these defects may manifest with hypercalciuria, which can cause nephrocalcinosis, lithiasis and renal failure, as well as other alterations specific to each syndrome. Hereditary hypophosphatemic rickets with hypercalciuria is a genetic disorder characterized by a loss of function of one of the sodium-phosphate cotransporter subtypes at renal level.32 The disorder manifests with high calcitriol levels due to a preserved calcitriol synthesis response to decreased phosphate levels. Elevated calcium levels and the presence of hypercalciuria contribute to the diagnosis, together with genetic analysis. In X-linked recessive hypophosphatemia, or Dent's disease, the biochemical alterations are similar to those seen in hereditary hypophosphatemic rickets with hypercalciuria, though female carriers only present hypercalciuria.25,33 Fanconi syndrome is associated with a generalized defect in proximal tubule function resulting in other alterations including glucosuria, hypouricemia, aminoaciduria and metabolic acidosis secondary to bicarbonate loss. As in hereditary hypophosphatemic rickets with hypercalciuria, elevated calcitriol concentrations have been reported.15
Treatment of hypophosphatemiaThe treatment of hypophosphatemia depends on the underlying cause and on other factors such as chronicity, severity, clinical manifestations, the presence of hypercalcemia or hypocalcemia, and renal function. Overt symptoms of hypophosphatemia are rare, unless the serum levels fall to <2mg/ml. Serious symptoms such as muscle weakness or rhabdomyolysis usually manifest when phosphate levels fall to <1mg/dl.7
In acute hypophosphatemia with phosphate depletion, phosphate supplementation may be carried out via the oral or intravenous route. Oral replacement is safer, but absorption is unpredictable and can cause gastrointestinal adverse effects such as diarrhea. Intravenous supplementing corrects hypophosphatemia faster, but poses the risk of causing hypocalcemia, arrhythmias, ectopic calcifications and acute renal failure.34 Severe acute hypophosphatemia (<1mg/dl) with phosphate depletion usually requires intravenous treatment, particularly in patients admitted to intensive care. In these cases, doses ranging from 0.25 to 0.50mmol/kg of sodium or potassium phosphate can be administered over an 8–12h period to a maximum of 80mmol. Strict monitoring of calcium and phosphorus levels is necessary in order to reduce the risk of ectopic calcifications and other complications.35 In less severe cases, treatment may be provided in the form of oral phosphorus supplements at a dose of 1–4g daily.
Chronic hypophosphatemia results from gastrointestinal or renal phosphate losses. Correction of the cause of hypophosphatemia sometimes requires the suppression of phosphate binders, diuretics, or the correction of hypomagnesemia. In mild cases, increased dietary phosphorus intake (half a liter of skimmed cow's milk provides about 450mg of phosphorus) may be sufficient. Renal losses may be due to genetic diseases characterized by increased FGF23 activity, as in XLHR or OO.36 In these cases, oral phosphate therapy is indicated to correct bone anomalies and restore normal growth in children. Conventional treatment of disorders involving FGF23 includes high dose phosphate supplementing (2–4g daily) in fractionated doses, and calcitriol (0.25–2μg/day).37 In OO, the objective is the removal of the causal tumor.
Recently, a monoclonal antibody targeted to FGF23 called burosumab has been developed and found to be effective in application to different forms of genetic or acquired hypophosphatemia. Burosumab treatment at doses of up to 1mg/kg every 4 weeks increases tubular phosphate reabsorption, normalizes phosphorus levels and increases calcitriol.38
HyperphosphatemiaHyperphosphatemia is defined as serum phosphorus levels >4.5mg/dl (1.45mmol/l) in adults, and >7mg/dl (2.26mmol/l) in children, confirmed by two measurements.
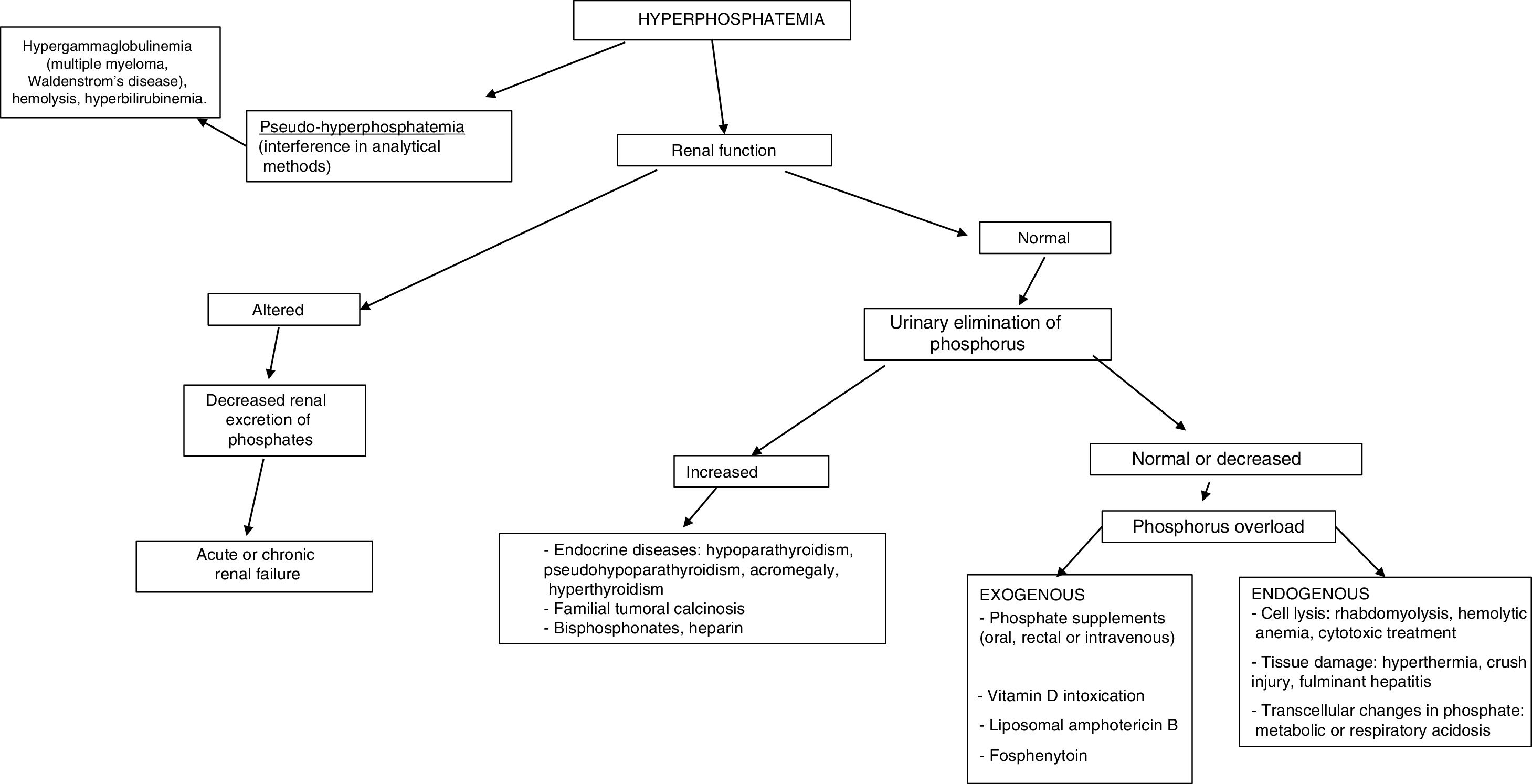
The most common cause of hyperphosphatemia is decreased renal phosphate excretion (Table 4), mainly due to acute renal failure or chronic kidney disease (CKD) in the presence of a glomerular filtration rate (GFR) <20ml/min/1.73m2. It may sometimes be caused by increased tubular reabsorption, redistribution from the intracellular to the extracellular compartment, or increased supply. Spurious hyperphosphatemia or pseudo-hyperphosphatemia is attributable to interference with the analytical methods: hyperglobulinemia, hyperlipidemia, hemolysis and hyperbilirubinemia, treatment with high doses of liposomal amphotericin B, sample contamination with recombinant tissue plasminogen activator, or heparin.
Causes of hyperphosphatemia.
| Altered renal phosphate excretion |
| Chronic renal failure (glomerular filtration rate<20ml/min/1.73m2) |
| FGF23 decrease: familial and non-familial tumoral calcinosis |
| Endocrine disorders: acromegaly, hypoparathyroidism and pseudohypoparathyroidism |
| Magnesium deficiency |
| Milk and alkaline syndrome |
| Drugs: heparin, bisphosphonates |
| Intracellular phosphate elevation |
| A. Increased supply (intravenous, oral, rectal) |
| Phosphate salts (oral or rectal laxatives) |
| Fosphenytoin |
| Liposomal amphotericin B |
| B. Phosphate transcellular changes: metabolic or respiratory acidosis |
| C. Catabolism or rapid cell lysis |
| Catabolic states |
| Tissue damage: hyperthermia, crush injury, fulminant hepatitis |
| Cell lysis: hemolytic anemia, rhabdomyolysis, cytotoxic treatment, severe leukemia |
Hyperphosphatemia is usually mild and asymptomatic; however, chronic hyperphosphatemia is an important factor in the development of secondary hyperparathyroidism in CKD. In severe acute hyperphosphatemia, the clinical manifestations arise from hypocalcemia caused by the formation of insoluble calcium phosphate salts: musculoskeletal weakness, tetany and increased neuromuscular excitability. At the central nervous system level seizures and cognitive impairment may develop.
The clinical manifestations of chronic hyperphosphatemia are related to the location of ectopic soft tissue calcification, and comprise pruritus, tendon rupture, band keratopathy and vascular calcification. These can be found in small arterioles and capillaries (calciphylaxis), causing necrotic skin lesions and subungual bleeding in medium-caliber arteries. Calcification may result in acute coronary syndrome (ACS) and cardiac arrhythmias, prolongation of the QT interval, or valve disease.3,39
Chronic hyperphosphatemia has been associated with increased mortality in patients with CKD on predialysis: there is a 35% increase in mortality per milligram increase in blood phosphorus concentration above the normal values. Other studies have not observed this association, however. In patients with CKD on dialysis, the mortality risk increases 18% for each milligram increase above the normal values.40
In establishing the etiological diagnosis, it is advisable to measure serum 25-hydroxyvitamin D, calcitriol, PTH, calcium corrected for albumin, magnesium, creatinine, urea, alkaline phosphatase, serum pH, and urine creatinine, calcium and phosphorus.3 A fractional excretion of phosphorus in urine <5% indicates a renal origin, while higher values are indicative of excess supply (Fig. 3).

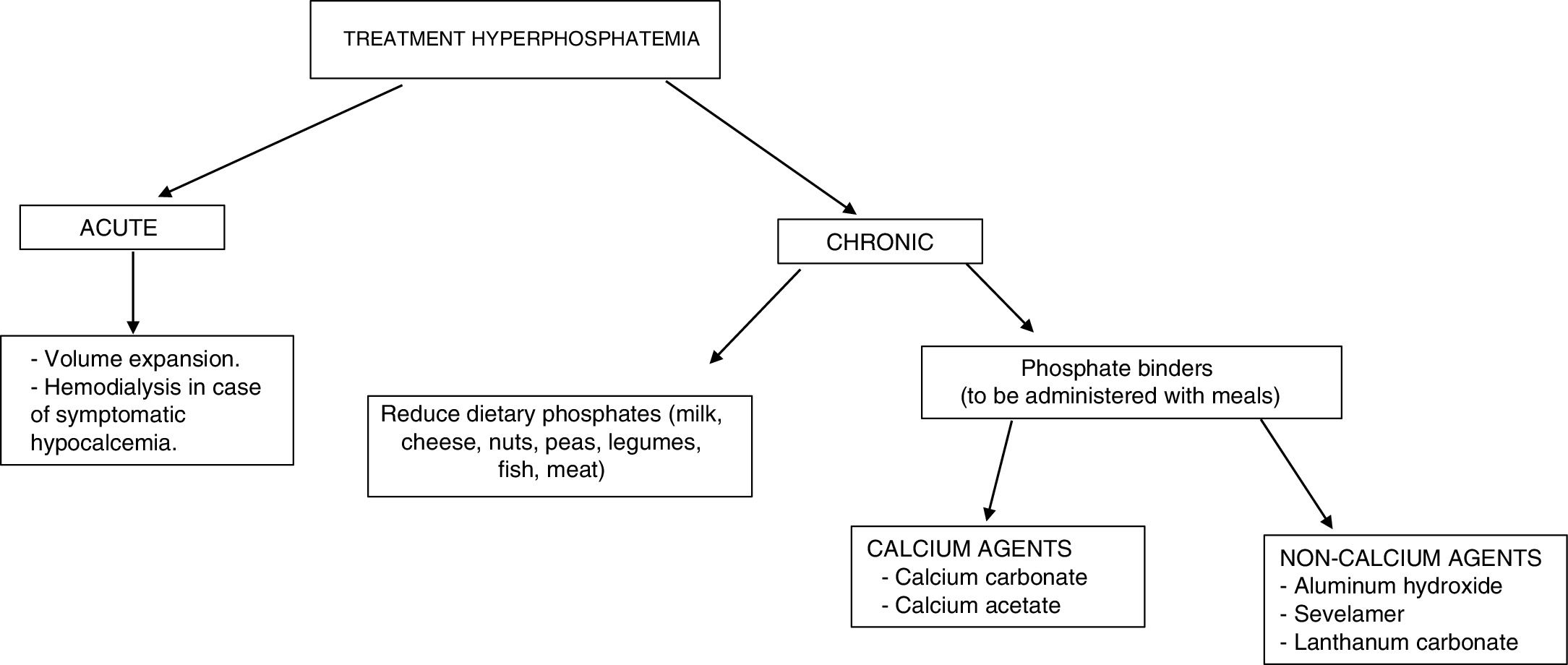
The treatment of hyperphosphatemia depends on its cause, the rapidity of onset, and the presence or absence of renal failure (Fig. 4).

Acute hyperphosphatemia.3,39 The treatment options are limited: volume expansion may be useful if renal function is preserved. It is essential to identify and suspend any exogenous source of phosphate; aluminum hydroxide antacids reduce its intestinal absorption and favor the binding of phosphate secreted by the intestine. Hemodialysis is the most effective treatment for hyperphosphatemia, and should be considered in severe cases characterized by acute onset.
Chronic hyperphosphatemia. In patients with CKD on predialysis, the treatment of progressive or persistent hyperphosphatemia is indicated.40 A restriction of dietary phosphorus intake is recommended (900mg/day), with the use of binding agents if hyperphosphatemia persists. Combination therapy may be started in patients with very high levels (>6mg/dl). Phosphorus binders cause a slight decrease in phosphorus levels in blood and in 24-h urine.41 The decrease in phosphorus concentrations in these patients has not been shown to improve important clinical parameters such as mortality or the progression of CKD.
In patients with CKD on dialysis, phosphorus intake restriction to 900mg/day is recommended, with or without associated phosphate binders.40 The restriction of phosphorus intake has not been shown to increase survival in these patients.42 The use of binders (chelating agents) has been associated with a 25–29% decrease in global mortality43,44 and a 22% decrease in cardiovascular mortality14 after a maximum of three years of follow-up.
The available chelating agents comprise carbonate and calcium acetate (calcium binders), and sevelamer and lanthanum (non-calcium binders). The use of non-calcium binders is associated with a 22–44% decrease in mortality compared with the use of calcium binders.45,46 This second group has been associated with hypercalcemia, adynamic bone disease and vascular calcifications.41 The choice of phosphate binder should be individualized: the clinical condition of the patient must be taken into account, as well as the cost of treatment, individual tolerability, and other mineral metabolic parameters such as PTH and calcemia.40
Conflicts of interestThe authors declare that they have no conflicts of interest.
Please cite this article as: García Martín A, Varsavsky M, Cortés Berdonces M, Ávila Rubio V, Alhambra Expósito MR, Novo Rodríguez C, et al. Trastornos del fosfato y actitud clínica ante situaciones de hipofosfatemia e hiperfosfatemia. Endocrinol Diabetes Nutr. 2020;67:205–215.
