




Los adenomas hipofisarios (AH) son los tumores selares más frecuentes y la causa más común de enfermedad hipofisaria1. Sin embargo, una amplia variedad de tumores no adenomatosos, metástasis, lesiones quísticas, infecciosas, vasculares o inflamatorias pueden afectar a la región selar2.
Los sarcomas son un grupo heterogéneo de tumores malignos que derivan de las células mesenquimales y crecen a partir del tejido conjuntivo esquelético y extraesquelético3. Se dividen en dos grandes grupos: sarcomas de partes blandas (SPB) y sarcomas óseos (SO)3. Tienen una prevalencia de cuatro a cinco casos (SPB) y un caso (SO) por cada 100.000 habitantes y representan el 1% de las enfermedades malignas en adultos y 12% en los niños4. Estos tumores pueden aparecer en cualquier parte del organismo, y tanto los SPB (angiosarcoma, liposarcoma, fibrosarcoma, sarcoma pleomórfico indiferenciado, leiomiosarcoma, rabdomiosarcoma) como los SO (sarcoma de Ewing, condrosarcoma, osteosarcoma) se han reportado afectando a la región selar5.
Presentamos el caso de un varón que, en 1993, a la edad de 42 años, se diagnosticó de una masa selar de forma incidental en una tomografía computarizada realizada por traumatismo craneoencefálico. No tenía síntomas o signos de disfunción hormonal. El estudio hipofisario basal, cortisol libre urinario de 24 horas y el test de ACTH-cortisol (Synacthen 250 μg) fueron normales. La resonancia magnética (RM) hipofisaria mostró una lesión selar con extensión supraselar e infiltración de ambos senos cavernosos. La campimetría reveló una hemianopsia bitemporal, por lo que se realizó craneotomía bifrontal con resección subtotal del tumor. El estudio histológico mostró una tumoración de 30 x 40 mm con inmunoreacción para ACTH, citoqueratina AE1/AE3, negativa para el resto de las hormonas hipofisarias con Ki67 < 2%, compatible con corticotropinoma silente. El estudio posquirúrgico mostró hipogonadismo hipogonadotrópico e hipotiroidismo secundario iniciando tratamiento hormonal sustitutivo. El paciente fue reintervenido por crecimiento tumoral en 1995 y 1998. La tumoración extirpada en ambas intervenciones tenía un patrón histológico similar con inmunoreacción para ACTH. Posteriormente, el paciente recibió radioterapia estereotáxica fraccionada (52 Gy). Un año después, la RM hipofisaria mostraba un resto tumoral quístico con diámetro máximo de 3 cm. En controles periódicos, el paciente permaneció clínica y radiológicamente estable durante 20 años.
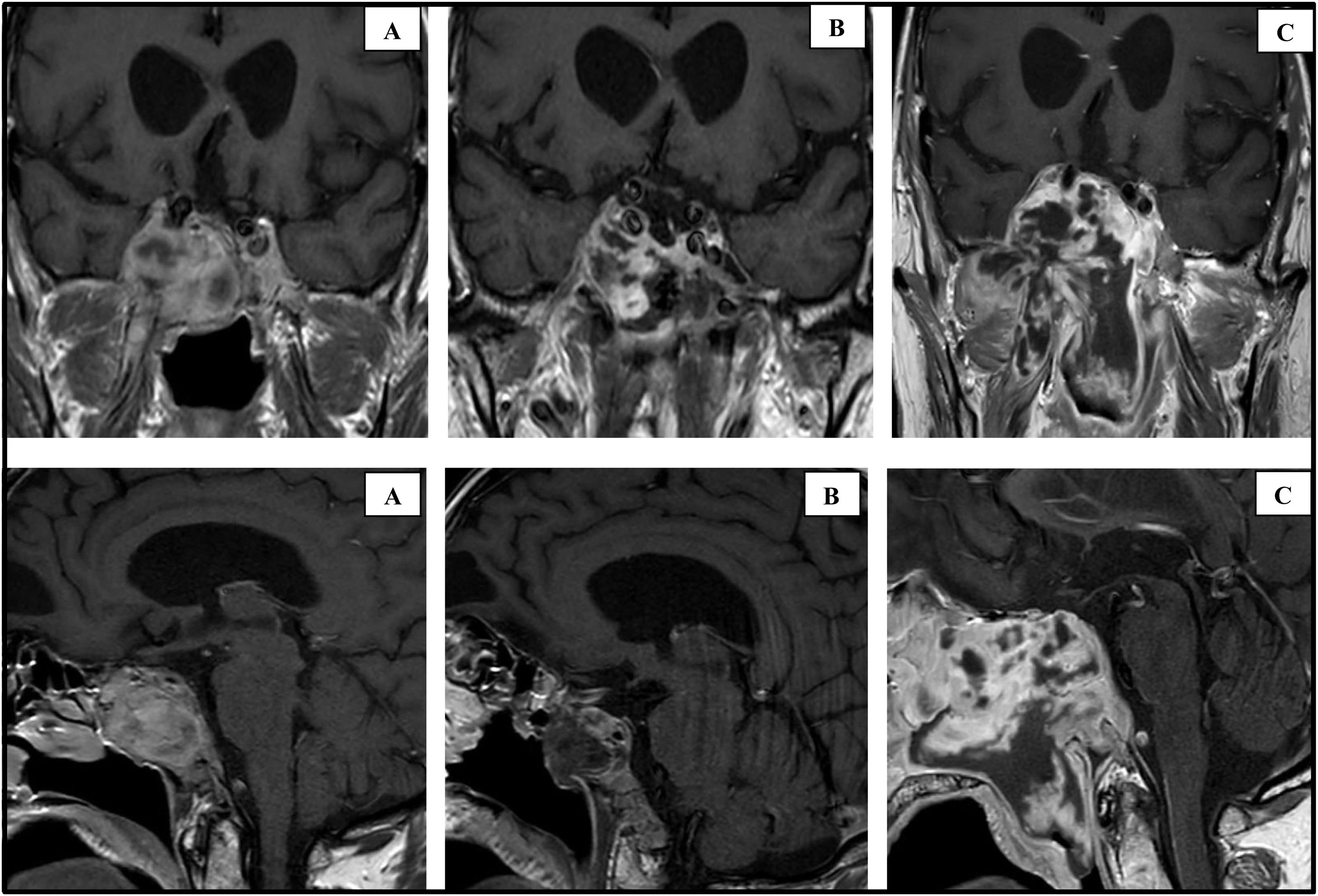
En octubre de 2019, a la edad de 69 años, el paciente consultó por cefalea, alteración visual y vómitos iniciados tres semanas antes. La exploración física evidenció parálisis del VI par craneal izquierdo. El estudio hormonal mostró GH sérica 0,02 μg/L (0-3,5); IGF-1 3,6 nmol/L (6,2-24,9); TSH < 0,01 mUI/L (0,57-5,51); T4L 15,8 pmol/L (12-22); prolactina 103 mUI/L (98-456); FSH 1,4 UI/L (1,5-12,4); LH < 1 UI/L (1,7-8,6); testosterona 8,4 nmol/L (6,7-25,7); testosterona libre 75 pmol/L (> 220); cortisol libre urinario 540 nmol/día (86-631); ACTH plasmática 1,9 pmol/L (2-12); cortisol sérico 284 nmol/L (172-497) con ascenso tras estimulación (Synacthen 250 μg) hasta 710 nmol/L. La RM hipofisaria evidenció un crecimiento de la lesión residual (fig. 1A). Se realizó resección parcial del tumor mediante cirugía transesfenoidal y el estudio histológico mostró inmunoreacción positiva difusa para vimentina, focal para actina, citoqueratina AE1/AE3 y negativa para desmina, CD34 y S100. El diagnóstico histológico fue sarcoma fusocelular con áreas de proliferación fibroblástica y condroblástica. La RM una semana después de la cirugía mostró reducción del tamaño de la lesión (fig. 1B) y el paciente fue dado de alta sin complicaciones inmediatas. Sin embargo, ocho semanas después consultó nuevamente por cefalea y alteración visual. Se constató en la RM hipofisaria un considerable crecimiento del tumor (fig. 1C), por lo que el paciente fue derivado a Oncología, donde inició tratamiento quimioterápico paliativo con doxorubicina, gemcitabina y dacarbazina; a pesar de lo cual falleció a los siete meses del diagnóstico del sarcoma.
 RM realizada en octubre de 2019 que muestra masa de 38 x 43 x 30 mm con infiltración del suelo selar, clivus, hueso occipital, comprensión del quiasma, nervio óptico derecho y ambos senos cavernosos. B) RM realizada una semana después de la última cirugía (noviembre de 2019), que muestra reducción de tamaño hasta 15 x 13 x 23 mm. C) RM realizada dos meses después de la última cirugía (enero de 2020), que muestra progresión del tumor hasta 66 x 74 x 66 mm.")
Cortes coronal y sagital de RM hipofisaria con gadolinio en diferentes momentos de la enfermedad.
A) RM realizada en octubre de 2019 que muestra masa de 38 x 43 x 30 mm con infiltración del suelo selar, clivus, hueso occipital, comprensión del quiasma, nervio óptico derecho y ambos senos cavernosos. B) RM realizada una semana después de la última cirugía (noviembre de 2019), que muestra reducción de tamaño hasta 15 x 13 x 23 mm. C) RM realizada dos meses después de la última cirugía (enero de 2020), que muestra progresión del tumor hasta 66 x 74 x 66 mm.
La incidencia de los sarcomas selares es desconocida y se han reportado menos de 100 casos hasta la actualidad5. Un estudio etiológico de 1.367 pacientes con masas selares/paraselares en la RM encontró tres sarcomas, lo cual representa el 0,2%6. Estos tumores se detectan en pacientes con edad media de 40 años y ligero predominio de varones5. Aunque la proporción de sarcomas inducidos por radioterapia es menor con las técnicas actuales7, el antecedente de radiación está presente en el 3-6% de los sarcomas y alcanza el 32% en los sarcomas que afectan la región selar5. El tiempo medio entre la radioterapia y la aparición del tumor es de 10 años, con rango muy variable (3-39 años)5. Los SPB selares están relacionados con mayor frecuencia a la radioterapia que los SO y los subtipos más frecuentes son el fibrosarcoma y el sarcoma pleomórfico indiferenciado5. En este sentido, el estudio histológico de nuestro paciente mostró un sarcoma muy poco diferenciado con una combinación de proliferación fibroblástica (SPB) y condroblástica (SO), que es poco común.
La causa de los sarcomas es desconocida, sin embargo, el factor ambiental de mayor fuerza etiológica es la radioterapia8. El desarrollo de estos tumores podría estar influido por dosis elevadas de radiación, exposición a edades tempranas y uso concomitante de ciertos quimioterápicos8. Algunos casos se han encontrado en el contexto de enfermedades hereditarias como el síndrome de Li-Fraumeni, neurofibromatosis tipo 1, síndrome de McCune-Albright o enfermedades de origen incierto, como la encondromatosis múltiple5,8. Se han relacionado también con virus como el herpes virus humano 8, el virus de Epstein-Barr o el VIH, los dos últimos reportados en pacientes con sarcomas de la región selar5,8.
Los sarcomas selares son tumores voluminosos (> 3 cm), que se presentan con síntomas de ocupación de espacio5. No hay datos clínicos, analíticos o radiológicos que permitan sospecharlos, por lo que son confundidos con AH no funcionantes u otros tumores no adenomatosos5. En nuestro caso, se consideró que el cambio de comportamiento del resto tumoral podría estar relacionado con la agresividad que pueden tener algunos corticotropinomas silentes. Sin embargo, el largo período de estabilidad previo y el antecedente de radioterapia nos orientaron a una transformación sarcomatosa como causa más probable. La resección quirúrgica completa es la única opción curativa, sin embargo, esta se consigue en menos del 20% de los casos4,5. La radioterapia y la quimioterapia se han empleado como opciones paliativas en tumores de crecimiento progresivo o enfermedad metastásica avanzada4,5, no obstante, la supervivencia media de estos pacientes es de seis meses5.
En conclusión, los sarcomas selares son tumores poco frecuentes, agresivos y de mal pronóstico que aparecen en pacientes de mediana edad. Es muy difícil su presunción antes de la cirugía, sin embargo, el cambio de comportamiento clínico y radiológico de un remanente tumoral previamente irradiado podría ser de utilidad en algunos casos. La extirpación completa es la única opción de supervivencia para estos pacientes.
FinanciaciónEste trabajo no ha recibido ningún tipo de financiación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
