




X-linked adrenoleukodystrophy (X-ALD) is a peroxisomal disorder affecting particularly the nervous tissue and adrenal cortex. Adrenomyeloneuropathy (AMN) is the most frequent phenotype, although adrenal insufficiency is usually the first manifestation in male patients. We set out to describe the clinical and biochemical features, together with the clinical course of X-ALD patients, focusing particularly on endocrine dysfunction.
Patients and methodsA retrospective study of 10 male X-ALD patients followed up at the Endocrinology Department. Epidemiologic data, phenotype evolution, endocrine and neurological findings and family history were analysed.
ResultsAll the patients presented with adrenal insufficiency, 4 of them during adulthood, with a mean age of 19.6±17.1 years (6–64 years). Six patients had mineralocorticoid deficiency. At diagnosis, 8 patients had Addison-only phenotype and 2 AMN phenotype. In the course of follow-up (24.9±16.1 years), 4 patients developed AMN about 25.0±7.4 years after the initial diagnosis and 2 patients presented the cerebral adult form 11 and 17 years after the initial diagnosis. Testosterone levels were within the normal range in all patients. There were 7 families, and age of onset and clinical course were similar in 3 of them.
ConclusionsThe presentation of X-ALD varied widely, 40% of the patients presented with adrenal insufficiency in adulthood, 60% had mineralocorticoid deficiency, and the onset and progression of neurological manifestations showed no pattern. Nevertheless, some similarities in the clinical course were found in some families. Our findings reinforce the need for screening for X-ALD at any age when approaching adrenal insufficiency and the importance of a multidisciplinary approach between endocrinologists and neurologists.
La adrenoleucodistrofia ligada al cromosoma X (X-ALD) es un trastorno peroxisomal que afecta especialmente al tejido nervioso y a la corteza suprarrenal. La adrenomieloneuropatía (AMN) es el fenotipo más común; no obstante, la insuficiencia suprarrenal generalmente es la primera manifestación en varones. Nuestro objetivo fue describir las características y el desarrollo clínico de pacientes con X-ALD, centrándonos en la disfunción endocrina.
Pacientes y métodoEstudio retrospectivo de 10 varones con X-ALD. Fueron analizados los datos epidemiológicos, evolución, hallazgos endocrinos, neurológicos e historia familiar.
ResultadosTodos los pacientes presentaron insuficiencia suprarrenal, 4 durante la edad adulta, con una edad media de 19,6±17,1años (6-64 años). Seis pacientes tenían deficiencia de mineralocorticoides. Al diagnóstico, 8 tenían insuficiencia suprarrenal aislada y 2 AMN. Durante el seguimiento (24,9±16,1), 4 desarrollaron AMN, 25,0±7,4años después del diagnóstico inicial y 2 presentaron la forma cerebral a los 11 y 17 años después del diagnóstico inicial. Los niveles de testosterona estaban dentro del rango normal en todos. Había 7 familias y la edad al diagnóstico y el desarrollo clínico fue similar en 3 de ellas.
ConclusionesLa presentación del X-ALD varió ampliamente, el 40% presentaron insuficiencia suprarrenal en la edad adulta, el 60% deficiencia de mineralocorticoides y el comienzo y progreso de las manifestaciones neurológicas no mostró ningún patrón. Sin embargo, unas pocas similitudes en el desarrollo clínico fueron encontradas en algunas familias. Nuestros resultados refuerzan la necesidad de excluir X-ALD a cualquier edad cuando se aborda la insuficiencia suprarrenal y la importancia de un enfoque multidisciplinar entre endocrinólogos y neurólogos.
X-linked adrenoleukodystrophy (X-ALD) is the most frequent peroxisomal disorder, with an estimated incidence of 1 in 16,800 new-borns (male and female).1,2 It is caused by mutations in the ABCD1 gene, resulting in the absence or dysfunction of adrenoleukodystrophy protein (ALDP), a peroxisomal transmembrane protein involved in the transmembrane transport of very-long-chain fatty acids (VLCFA). This disorder is characterized by an impaired peroxisomal beta-oxidation of VLCFA, leading to their accumulation in plasma and in all tissues, particularly affecting the nervous tissue and adrenal cortex.3,4
The exact pathophysiological mechanisms by which the excess of VLCFA leads to tissue damage and why specific cell types are uniquely vulnerable are not fully understood. However, direct VLCFA cytotoxicity and the induction of oxidative stress are both known to play a role.3,5–7
The clinical manifestations are highly variable and there is no correlation between genotype and phenotype, even within individual families.8 Phenotypes in men range from asymptomatic and pre-symptomatic patients, Addison-Only (ADO) and Adrenomyeloneuropathy (AMN) to the cerebral form of X-ALD (CALD), including childhood, adolescent and adult forms.9 However, phenotypes are not static, and X-ALD is considered a progressive disease and the risk of presenting symptoms increases with age.6,7
Women with X-ALD are also affected and are not just carriers. More than 80% of women with X-ALD develop myelopathy or peripheral neuropathy after the age of 60 years, although adrenal insufficiency or cerebral involvement are very rare.10
Most X-ALD reports have focused on the neurological manifestations of this metabolic disease. However, endocrine dysfunction is common, and the first manifestation in male patients is usually adrenal insufficiency. Almost all males with X-ALD develop adrenal insufficiency in the course of their life and about 80% before adulthood.11 Gonadal dysfunction may also be present in X-ALD, although it is not always clinically apparent.12
In this study, we describe the clinical and biochemical features, together with the clinical course of 10 X-ALD patients, focusing not only on the neurological alterations, but also on the endocrine dysfunctions associated with X-ALD.
Materials and methodsWe performed a retrospective study, including 10 patients diagnosed with X-ALD and followed up at the Endocrinology Department of Hospital de Santa Maria, Centro Hospitalar Universitário Lisboa Norte (Lisbon, Portugal).
Information was collected through their medical records, including demographics, endocrine and neurological findings, neuroimaging and biochemical data, treatment and disease progression. The data collected included gender, X-ALD phenotype, date of birth and current age/date of death, age at diagnosis, brain MRI data and testosterone levels, phenotype evolution, comorbidities, treatment schemes and family history.
ResultsX-ALD presentation and clinical courseThis retrospective study included a total of 10 male patients with a mean age of 19.6±17.1 years (range: 6–64 years) at diagnosis who were followed up on average for 24.9±16.1 years. The patients belong to 7 different families, and all of them except two had a positive family history for X-ALD (Table 1).
General characteristics of the 10 patients with X-ALD.
| Patient | Age (years) | Age at diagnosis (years) | Adrenal insufficiency | Phenotype | Families | Follow-up (years) |
|---|---|---|---|---|---|---|
| 1 | 51 | 6 | + | AMN | 1 | 45 |
| 2 | 63 | 6 | + | AMN | 1 | 57 |
| 3 | 34 | 13 | + | ADO | 2 | 21 |
| 4 | 41 | 14 | + | ADO | 2 | 27 |
| 5 | 45 | 18 | + | AMN | 2 | 27 |
| 6† | 80 | 64 | + | CALD | 3 | 16 |
| 7 | 39 | 13 | + | CALD | 4 | 26 |
| 8 | 29 | 11 | + | ADO | 5 | 18 |
| 9 | 24 | 22 | + | ADO | 6 | 2 |
| 10 | 39 | 29 | + | AMN | 7 | 10 |
ADO: Addison-Only; AMN: adrenomyeloneuropathy; CALD: cerebral adrenoleukodystrophy; “+” present; †: deceased
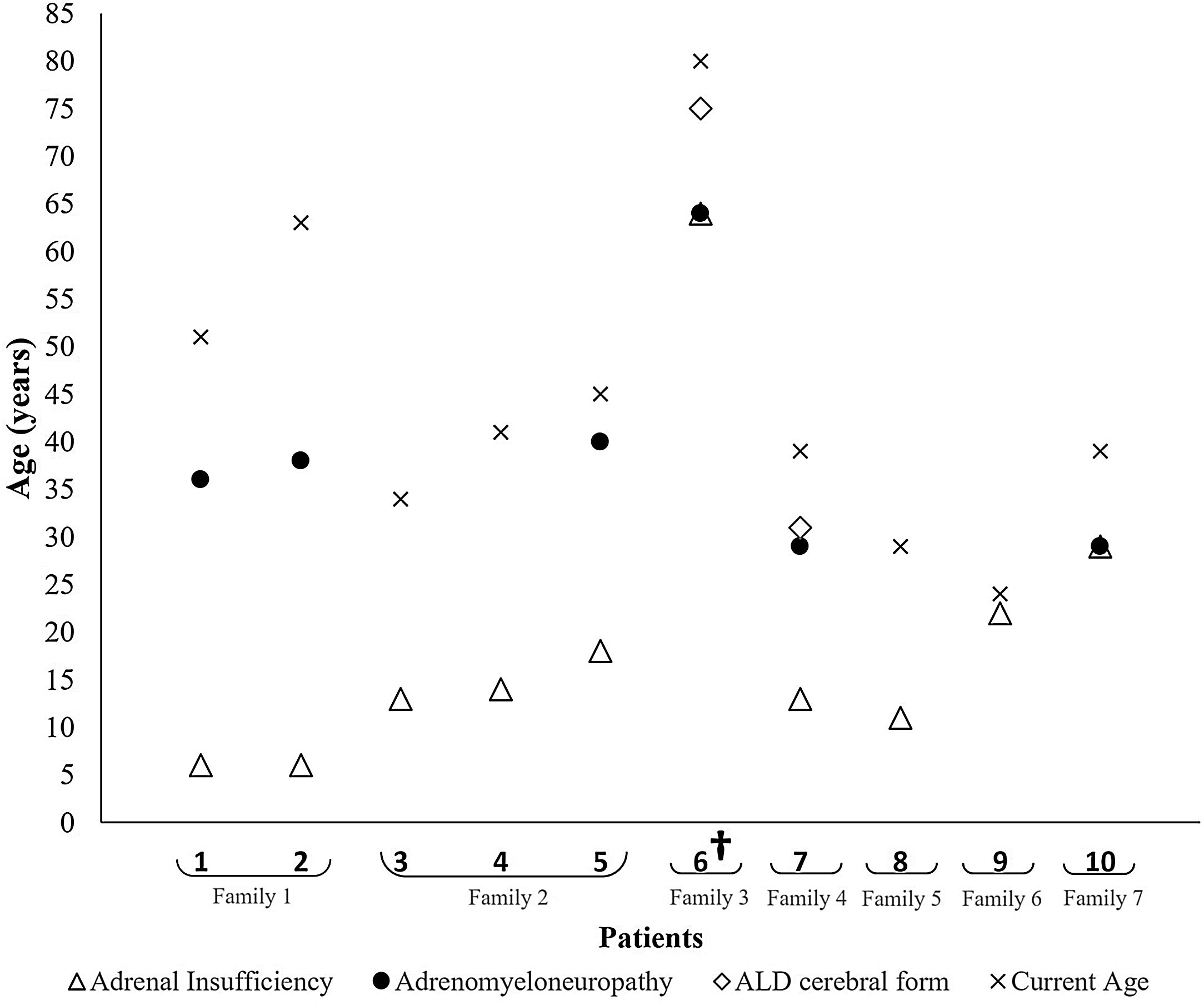
At diagnosis, all the patients presented adrenal insufficiency, 8 of them with ADO phenotype and the remaining 2 with AMN phenotype. The patients presenting with ADO phenotype had a mean age of 12.9±5.5 years, whereas those who presented with AMN phenotype had a mean age of 46.5±24.7 years (Table 1 and Fig. 1).

Fig. 1 shows the phenotype evolution of the 10 patients included. Throughout the follow-up, 4 additional patients also developed AMN, on average about 25.0±7.4 years after the initial diagnosis, ranging from 16 to 32 years. Overall, AMN was diagnosed at a mean age of 39.3±12.9 years. Two patients also presented the adult form of CALD at 75 and 30 years, 11 and 17 years after the initial diagnosis, respectively. The first patient died two months after this diagnosis and the second one remains in follow-up.
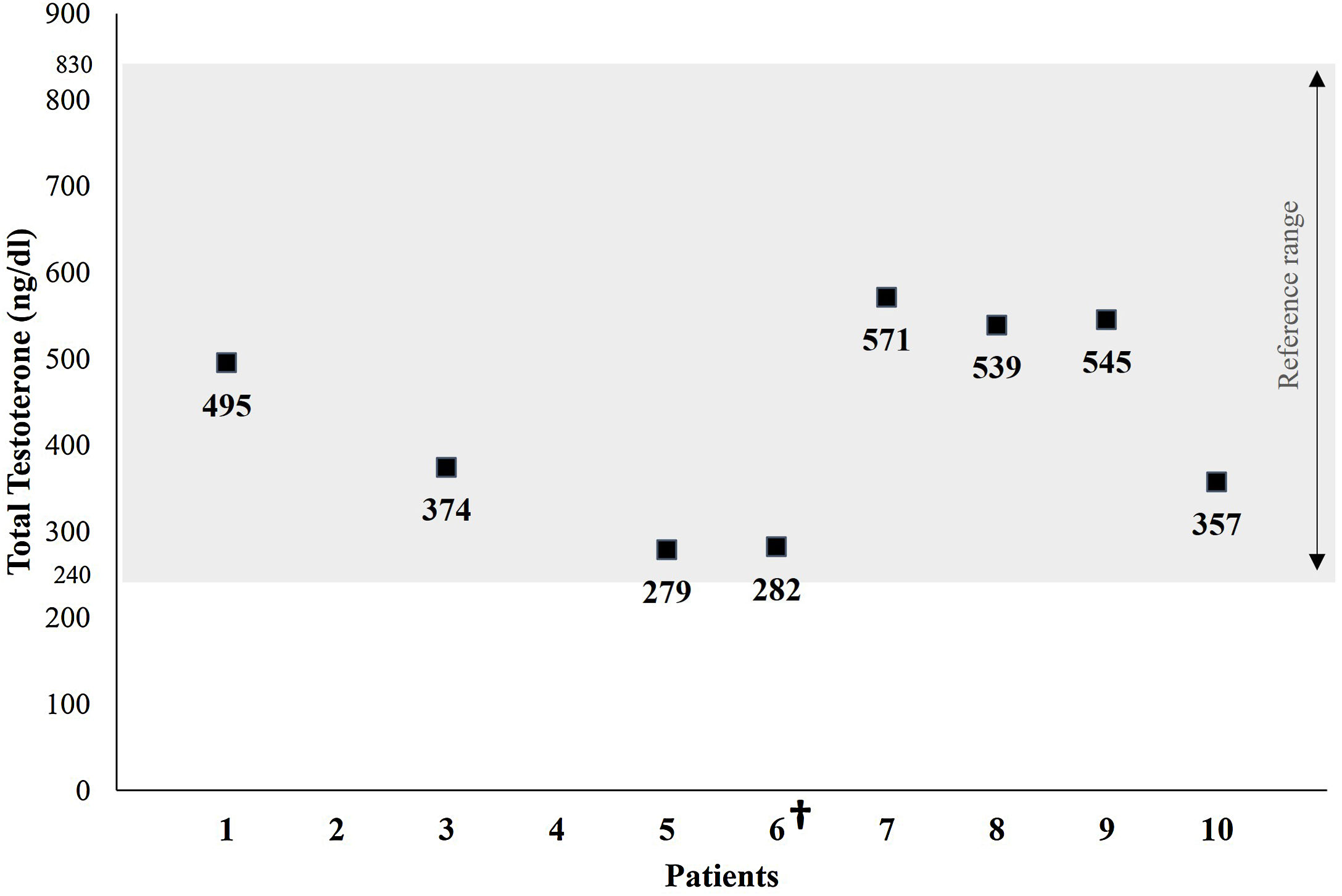
A single total testosterone determination was available in 8 of the patients studied. Testosterone levels were within the normal range in all the patients tested, with an average of 430.2±120.9ng/dL (reference range: 240–830ng/dL) (Fig. 2).
; the grey area represents the laboratory normal reference range for men; no data are available for patients 2 and 4; †: deceased.")
Two patients were also diagnosed with hypothyroidism, both of whose antithyroid peroxidase and antithyroglobulin antibodies were within the normal range.
As for neurological manifestations, all six patients with AMN presented spastic paraparesis and sensitive peripheral neuropathy, 5 mood disorders, 4 sphincter disturbances and 2 sexual dysfunction (Table 2).
Neurological manifestations of the 10 patients with X-ALD.
| Patient | Spastic paraparesis | Sensitive peripheral neuropathy | Mood disorders | Sphincter disturbances | Sexual dysfunction |
|---|---|---|---|---|---|
| 1 | + | + | − | + | − |
| 2 | + | + | − | + | − |
| 3 | − | − | − | − | − |
| 4 | − | − | + | − | − |
| 5 | + | + | + | − | − |
| 6† | + | + | + | + | − |
| 7 | + | + | + | + | + |
| 8 | − | − | − | − | − |
| 9 | − | − | + | − | − |
| 10 | + | + | − | − | + |
“+”: present; “−”: absent; †: deceased.
All the AMN patients exhibited slight brain MRI abnormalities, presenting with T2 and moderate hyperintensity (FLAIR) in the internal capsule area. In the CALD patients, patient number 7s brain MRI showed an increased signal on T2 and FLAIR sequences in the corpus callosum, pyramidal tracts within the brainstem, pons and internal capsules, together with enhancement on T1 sequences after intravenous gadolinium administration. Unfortunately, the brain MRI of patient 6 was not available.
Family historyAs was mentioned above, the ten patients belonged to seven different families, marked from 1 to 7 on Table 1 and Fig. 1.
Family 1 is comprised of two brothers, currently aged 51 and 63 years, both of whom presented adrenal insufficiency at the age of six and who developed AMN by the ages of 36 and 38 years, respectively.
Family 2 included two brothers and one maternal cousin, now aged 34, 41 and 45 years. All of them presented adrenal insufficiency at the ages of 13, 14 and 18 years, respectively. Only the third was diagnosed with AMN by the age of 40.
Family 4 is represented by one 39-year-old patient, diagnosed with adrenal insufficiency at the age of 13 years and who developed both AMN and cerebral involvement at the age of 29. Although he remained clinically stable for several years, he has presented clinical and imaging signs of disease progression for the last 2 years. His family history includes a brother with X-ALD who died with CALD at the age of 33 years.
Patients 6 and 8 represent families 3 and 5. The family history of the former revealed 3 brothers diagnosed with X-ALD, and the latter a maternal grandfather also diagnosed with X-ALD. However, information about phenotypes and clinical course was unavailable in their clinical records.
X-ALD treatmentA VLCFA-restricted diet was recommended for all of the patients accompanied. Lorenzo's oil was used by 3 patients, 2 with ADO phenotype and 1 with AMN (Table 3).
Therapeutic schemes of the 10 patients with X-ALD.
| Patient | Lorenzo's Oil | Glucocorticoid therapy | Glucocorticoid dose (mg/day) | Mineralocorticoid therapy | Fludrocortisone dose (mg/day) |
|---|---|---|---|---|---|
| 1 | − | Hydrocortisone | 15 | − | − |
| 2 | − | Hydrocortisone | 20 | − | − |
| 3 | + | Hydrocortisone | 15 | + | 0.1 |
| 4 | − | Hydrocortisone | 30 | − | − |
| 5 | + | Hydrocortisone | 15 | + | 0.1 |
| 6† | − | Prednisolone | 7.5 | + | 0.05 |
| 7 | − | Hydrocortisone | 15 | + | 0.1 |
| 8 | + | Hydrocortisone | 15 | + | 0.05 |
| 9 | − | Hydrocortisone | 25 | + | 0.1 |
| 10 | − | Hydrocortisone | 25 | − | − |
“+”: yes; “−”: no; †: deceased.
Hydrocortisone was used in 9 patients at an average daily dose of about 19.4±5.8mg. One patient was medicated with 7.5mg of prednisolone per day. Six patients were medicated with fludrocortisone at an average daily dose of approximately 0.08±0.03mcg (Table 3).
Allogeneic haematopoietic stem cell transplantation (HSCT) was not performed in any of the patients with CALD.
DiscussionX-ALD has traditionally been classified in different phenotypes, including ADO, AMN, childhood, adolescent or adult CALD and asymptomatic carriers. However, X-ALD is a progressive disease and the risk of having symptoms increases with age.5,6,9 New-born screening for X-ALD permits the detection of the disease prior to the development of clinical manifestations and has already been implemented by several countries.7
Typically, patients are born asymptomatic, and adrenal insufficiency is usually the first manifestation of the disease, affecting up to 80% of men with X-ALD.7,11 Here we have described a series of 10 X-ALD patients, all presenting with adrenal insufficiency at diagnosis, 80% of them with ADO phenotype.
Adrenal dysfunction in X-ALD is due to primary adrenocortical insufficiency. However, the nature of adrenal gland toxicity and its relationship with increased VLCFA levels is poorly understood. Nevertheless, abnormal VLCFA accumulation in the adrenal cortex, particularly evident in the zona reticularis and the zona fasciculata, is believed to cause apoptosis and ultimately adrenal cortex atrophy.3,7,13 Furthermore, the incorporation of VLCFA into the adrenocortical cell membrane may also impair adrenocorticotropin (ACTH) receptor function, and the increased esterification of cholesterol with VLCFA can lead to a relative shortage of substrate for steroidogenesis and impair cortisol secretion further.3,13,14
The accumulation of VLCFA has been detected in the foetal adrenal gland, indicating that these alterations are already present in utero. However, loss of adrenal function appears to be a gradual and progressive phenomenon, as subclinical abnormalities in cortisol response to ACTH stimulation usually precede frank hypocortisolism.7,13,15
Adrenal insufficiency is most commonly diagnosed between the ages of 3 and 10 years, although it has been reported as early as 5 months of age.11,13,16 However, in our study, only two patients were diagnosed before the age of 10 years and 4 patients were even diagnosed during adulthood, including one patient who was surprisingly diagnosed at the age of 64, illustrating the variability in the presentation of X-ALD.
Acknowledging that Addison's disease is due to X-ALD has important implications not only for genetic counselling but also for its management. It is therefore important to screen for X-ALD in male patients with primary adrenal insufficiency, particularly in those diagnosed before 10 years of age. Nevertheless, our results underscore the need to also include VLCFA assessment in adult patients with primary adrenal insufficiency, particularly in those with concomitant neurological symptoms.
As was already mentioned, X-ALD is a progressive disease, and the phenotypes are not static. Indeed, since AMN appears later in life and will eventually affect all men with X-ALD who reach adulthood, periodical neurological surveillance is recommended, even in patients presenting with ADO phenotype.3,16–18 In our series, AMN was diagnosed in half of the patients initially presenting with ADO. However, the progression rate was highly variable, as the time between the diagnosis of adrenal insufficiency and AMN development ranged from 16 to 32 years.
AMN is mainly diagnosed in the third and fourth decade of life, and its incidence increases with age.3–6 Spinal cord lesions, particularly the long tracts and to a lesser extent of the peripheral neural system, are observed in AMN, traditionally characterised as a distal axonopathy without significant myelin changes.9,19 Therefore, AMN symptoms represent a combination of myelopathy and peripheral neuropathy, gradually progressive, and most patients lose unassisted ambulation by the 6th decade.5,17 A higher prevalence of major affective disturbance has also been reported in AMN patients.20 In our series, the clinical presentation of the six AMN patients was in line with the traditional presentation and, remarkably, 4 of them also presented with mood disorders, although CALD was not apparent.
The cerebral form of ALD is the most rapidly progressive and devastating phenotype of X-ALD, characterised by a severe inflammatory demyelination process, affecting primarily the cerebral hemispheres.3,7,16,21 CALD presents more frequently during childhood, although it can emerge at any age and is believed to be the result of an interplay between genetic and environmental factors. The onset of CALD is usually insidious and it presents in a similar fashion in children and adults, although progression appears to be slower in adults.6,7,17,22 Of the two patients in our series who were diagnosed with the adult form of CALD, one of them passed away two months after the diagnosis. However, and remarkably so, the other patient is still alive, with mild neurological symptom progression 8 years on from the initial diagnosis.
There is no generalised correlation between genotype and phenotype in X-ALD, meaning that the clinical course is unpredictable, even in individual families.8,23 Nevertheless, we found certain similarities both in age of presentation and clinical course in 3 of the families studied in our case series. Therefore, it cannot be ruled out that certain mutations lead to specific phenotypes or influence the age of onset of X-ALD clinical manifestations, and further studies are needed to address this issue.
Male X-ALD patients may also present with hypogonadism, which is most usually seen once neurological or adrenal symptoms are present. Most commonly, patients present with primary gonadal failure, related to VLCFA accumulation on Leydig cells which seems to affect testicular function directly. However, androgen receptor dysfunction due to VLCFA accumulation has also been suggested.12,13
Testosterone levels are reported to be in the lower-normal range, and in a previously published study only 2.5% of the X-ALD patients were on testosterone replacement therapy due to primary hypogonadism.7,13,24 Although men with X-ALD have higher rates of testicular dysfunction than the general population, infertility rates were reportedly comparable between both.25 Furthermore, erectile dysfunction, a common finding among X-ALD patients, usually reflects neurological impairment rather than testosterone deficiency.12,13
In our series, none of the men with X-ALD presented with hypogonadism and no history of infertility was reported. However, we have only evaluated the last total testosterone determination available and it remains to be answered whether a longer follow-up period could have influenced our results.
There is currently no curative or preventive treatment for the majority of patients with X-ALD. For childhood CALD, HSCT, when performed in the early stages of cerebral involvement, has been shown to halt the progression of neurological disease and increase disease-specific survival.26 However, whether or not HSCT has any impact on the occurrence or progression on AMN of X-ALD remains to be seen.5,7 Autologous transplantation of genetically-modified hematopoietic stem cells is currently being explored as an alternative to HSCT, although the long-term efficacy of this therapy has yet to be determined.27 In our series, since the two patients with CALD were only diagnosed during adulthood, HSCT was not considered.
Lorenzo's oil, a mixture of oleic acid (C18:1) and erucic acid (C22:1), normalises plasma C26:0 levels within 1 month in most patients with ALD. However, C26:0 levels in the nervous system are not affected, and in several open-label trials, Lorenzo's oil failed to improve neurological or endocrine function and proved to be ineffective in halting AMN progression.28 Three patients in our series had been medicated with Lorenzo's oil, one of whom progressed to AMN at the age of 40, 22 years after the diagnosis of adrenal insufficiency.
The treatment of adrenal insufficiency in X-ALD is no different to that of individuals with other forms of primary adrenal insufficiency, in whom glucocorticoid replacement therapy is mandatory. Importantly, neither allogenic nor autologous HSCT can prevent or reverse adrenal insufficiency.29
Since AI in X-ALD is an evolving process, glucocorticoid replacement therapy may initially only be required during stressful times, although progressively daily glucocorticoid treatment will be needed. Glucocorticoid replacement requirements are generally the same as in other forms of primary AI.6,13,16 Due to the relative sparing of the zona glomerulosa from the accumulation of VLCFA, frank hypoaldosteronism with salt wasting is not frequent, and mineralocorticoid function often remains intact. Nevertheless, screening and treatment for mineralocorticoid deficiency should always be considered, since approximately between one third and one half of men with X-ALD ultimately present with impaired mineralocorticoid function.14,15
In our series, glucocorticoid replacement therapy was initiated in all patients at diagnosis, and 6 out of 10 patients were on mineralocorticoid replacement therapy (60%), a slightly higher proportion than the reports of previous studies.13
In males with X-ALD with confirmed testicular dysfunction, testosterone replacement therapy should be considered if clinically indicated. However, decreased libido and erectile dysfunction, common findings among X-ALD patients, usually reflect neurological impairment and/or the presence of a chronic disease rather than a testosterone deficiency.12,13 Similarly, although erectile dysfunction was reported in two patients in our series, none of the men with X-ALD presented with hypogonadism. The main limitations of our study relate to its retrospective nature and limited access to clinical records. Nevertheless, we were able to describe the main clinical features of ten patients with a rare metabolic disease, followed up for a considerable period.
In conclusion, we have described a series of 10 patients with X-ALD followed up at an Endocrinology Department, all of whom presented with adrenal insufficiency and none with hypogonadism. The presentation of adrenal insufficiency varied widely, with a 40% presentation rate during adulthood and a 60% rate of mineralocorticoid deficiency. Our findings reinforce the need to screen for X-ALD at any age when dealing with adrenal insufficiency, as well as the importance of regular screening for mineralocorticoid deficiency in X-ALD patients. Progression rate and age at presentation of neurological manifestations varied in our series, which strengthens the need for a multidisciplinary approach, including endocrinologists and neurologists, throughout follow-up.
Data availability statementAll the data generated or analysed during this study are included in this published article.
Authors’ contributionsAll the authors contributed to the conception and design of the study. Data collection and analysis were performed by TM. The manuscript was written by TM and CC and SV reviewed the manuscript and provided critical input. All the authors read and approved the final manuscript.
FundingThere was no funding involved in the development of this article.
Conflict of interestThe authors declare that they have no conflict of interest.
