









The term hypophysitis is used to designate a heterogeneous group of pituitary conditions characterized by the presence of inflammatory infiltration of the adenohypophysis, neurohypophysis, or both. Although hypophysitis are rare disorders, the most common in clinical practice is lymphocytic hypophysitis, a primary hypophysitis characterized by lymphocytic infiltration, which predominantly affects women. Other forms of primary hypophysitis are associated with different autoimmune diseases. Hypophysitis can also be secondary to other disorders such as sellar and parasellar diseases, systemic diseases, paraneoplastic syndromes, infections, and drugs, including immune checkpoint inhibitors. The diagnostic evaluation should always include pituitary function tests and other analytical tests based on the suspected diagnosis. Pituitary magnetic resonance imaging is the investigation of choice for the morphological assessment of hypophysitis. Glucocorticoids are the mainstay of treatment for most symptomatic hypophysitis.
El término hipofisitis se emplea para designar un conjunto heterogéneo de lesiones hipofisarias caracterizadas por una infiltración inflamatoria de la adenohipófisis, neurohipófisis o ambas. Aunque las hipofisitis son poco frecuentes, la forma clínica más común es la hipofisitis linfocítica, una forma de hipofisitis primaria caracterizada por infiltración linfocitaria, que afecta predominantemente a mujeres. Otras formas de hipofisitis primaria se asocian a distintas enfermedades autoinmunes. La hipofisitis puede ser también secundaria a otros trastornos, como lesiones selares y paraselares, enfermedades sistémicas, síndromes paraneoplásicos, infecciones y fármacos, entre los que destacan los inhibidores de los puntos de control inmunitarios. La evaluación diagnóstica debe incluir siempre pruebas de función hipofisaria y otras pruebas analíticas en función de la sospecha diagnóstica. La resonancia magnética hipofisaria es la prueba de elección para la valoración morfológica de la hipofisitis. Los glucocorticoides son la base del tratamiento de la mayoría de las hipofisitis sintomáticas.
Hypophysitis is defined as an inflammation of the pituitary gland which may be accompanied by different degrees of hormone deficiency. Depending on the extent of anatomical spread, the inflammation may affect the anterior pituitary (adenohypophysitis) (∼65%), the infundibulum and posterior pituitary (infundibuloneurohypophysitis) (∼10%) or the entire pituitary gland (panhypophysitis) (∼25%).1–6 On occasion, the involvement can extend to the hypothalamus or even affect it in isolation (hypothalamitis).6 Hypophysitis can be divided according to the aetiology into primary hypophysitis (autoimmune or idiopathic) and those secondary to different lesions affecting the sellar or parasellar area, systemic diseases, cancers, infections and drugs5–7 (Table 1). The secondary types of hypophysitis are more common (75.1%) than idiopathic inflammatory lesions (15.4%).8 Histopathologically, hypophysitis is classified into five types, which are lymphocytic, granulomatous, IgG4-related (IgG4-RH), xanthomatous and necrotising, although there are also mixed forms5,6 (Table 2).
Aetiological classification of hypophysitis.
| Primary |
| Isolated |
| Lymphocytic hypophysitis |
| Associated with autoimmune diseases |
| Autoimmune polyglandular syndromes |
| Autoimmune thyroiditis |
| Autoimmune adrenalitis |
| Type 1 diabetes mellitus |
| Lymphocytic parotitis |
| Idiopathic inflammatory myopathy |
| Systemic lupus erythematosus |
| Sjogren's syndrome |
| Rheumatoid arthritis |
| Primary biliary cirrhosis |
| Atrophic chronic gastritis |
| Optic neuritis |
| Myocarditis |
| Temporal arteritis |
| Behcet's disease |
| Retroperitoneal fibrosis |
| Erythema nodosum |
| Idiopathic thrombocytopenic purpura |
| Dacryoadenitis |
| Autoimmune thrombocytopenia |
| Autoimmune encephalitis |
| Secondary |
| Sellar/parasellar diseases |
| Germinoma |
| Rathke's pouch cyst |
| Craniopharyngioma |
| Pituitary adenoma |
| Pituitary apoplexy |
| Glioma |
| Meningioma |
| Pituicytoma |
| Chordoma |
| Teratoma |
| Pituitary lymphoma |
| Systemic diseases |
| IgG4-related disease |
| Sarcoidosis |
| Granulomatosis with polyangiitis |
| Langerhans cell histiocytosis |
| Erdheim-Chester disease |
| Rosai-Dorfman disease |
| Inflammatory pseudotumour |
| Tolosa-Hunt syndrome |
| Takayasu arteritis |
| Cogan's syndrome |
| Crohn's disease |
| Paraneoplastic syndromes |
| Anti-PIT1 antibody syndrome |
| Isolated ACTH deficiency |
| Infections |
| Bacterial (Mycobacterium tuberculosis, Treponema pallidum, Tropheryma whipplei, Borrelia, Brucella) |
| Viral (SARS-CoV-2, cytomegalovirus, herpes simplex, varicela zoster, influenza, coronavirus, enterovirus, Coxsackie) |
| Mycotic (Aspergillus, Nocardia, Candida albicans, Pneumocystis jirovecii) |
| Parasitic (Toxoplasma gondii) |
| Drugs |
| Immune checkpoint inhibitors (anti-CTLA-4, anti-PD1 and anti-PD-L1) |
| Interferon-α |
| Ribavirin |
| Ustekinumab |
Adapted and modified from Prete and Salvatori.5
Characteristics of hypophysitis according to its histopathological classification.
| Lymphocytic | Granulomatous | IgG4-related | Xanthomatous | Necrotising | |
|---|---|---|---|---|---|
| Prevalencea (%) | 68 | 19 | 8 | 4 | <1 |
| Age (years) | 30 (female) | 44 | 70 (male) | 40 | 33 |
| 40 (male) | 20−30 (female) | ||||
| Female:male ratio | 3:1 | 3:1 | 1:2 | 3:1 | 1:2 |
| Related to pregnancy | 70% related to pregnancy or postpartum | No | No | No | No |
| Histopathology | Diffuse infiltration of lymphocytes (mainly T cells) in the pituitary. Occasional lymphoid follicles, plasma cells, eosinophils and fibroblasts. Fibrosis and pituitary atrophy may occur late in the disease | Large numbers of multinucleated giant cells and histiocytes with granuloma formation | Extensive glandular infiltration by plasma cells with a high degree of IgG4 positivity. Storiform fibrosis is seenb. Fibrosis and pituitary atrophy occur in later stages of the disease if left untreated | Foamy histiocytes (lipid-rich macrophages) without the presence of granulomas. Plasma cells and small round mature lymphocytes can also be seen. Pituitary fibrosis can be seen in later stages of the disease | Diffuse nonhaemorrhagic necrosis with surrounding lymphocytes, plasma cells and eosinophils |
Adapted and modified from Prete and Salvatori.5
The diagnosis of hypophysitis is based on clinical, laboratory, and radiological data. Although pituitary biopsy is the definitive test for the diagnosis of primary hypophysitis, it is not routinely performed and is usually reserved only for selected cases, in which decompressive surgical treatment is required, or in which the specific histopathological diagnosis changes the approach.6,9 Clinical suspicion is therefore based on the presence of hypopituitarism with or without central diabetes insipidus, combined with pituitary gland enlargement visible on magnetic resonance imaging (MRI), which may or may not also be accompanied by headache and, in some cases, compression of the optic pathways and/or cranial nerves, with the corresponding neuro-ophthalmological signs. However, as we discuss later, a normal MRI does not rule out a suspected diagnosis, particularly in cancer patients being treated with immune checkpoint inhibitors (ICPI) and in the late phases of this process. As hypophysitis may be secondary to a systemic disease, a detailed medical history, physical examination and investigations aimed at ruling out these possible alternative diagnoses are important. It is essential to review all the medication the patient takes, particularly in cancer cases, especially those treated with immunotherapy.
Guide to diagnosisAnalytical studiesHormonal and biochemical studiesIn the diagnostic assessment, all the anterior pituitary axes should be studied by means of a baseline study that includes cortisol, corticotropin (ACTH), thyrotropin (TSH), free thyroxine, prolactin, luteinising hormone (LH), follicle-stimulating hormone (FSH), oestradiol in premenopausal women, testosterone in males and insulin-like growth factor 1.6,10 Attention should also be paid to urinary volume, electrolytes, and serum and urine osmolality, as hyponatraemia may occur from secondary adrenal insufficiency, severe hypothyroidism, or syndrome of inappropriate secretion of antidiuretic hormone (SIADH), or hypernatraemia and dehydration in cases of central diabetes insipidus.6,11
Low- or high-dose ACTH stimulation testing may be necessary for the diagnosis of secondary adrenal insufficiency, but we have to remember that this test is not designed for the diagnosis of secondary adrenal insufficiency, as it may not detect a recent-onset ACTH deficiency. The diagnosis of growth hormone (GH) deficiency also requires stimulation tests, but it is not necessary if there are three hormone axes affected. Prolactin levels may be decreased in hypophysitis due to deficiency or increased due to compression of the stalk. Assessment of neurohypophysis function is necessary in patients with polyuria and may require a fluid restriction test or copeptin determination.12 Central diabetes insipidus may be masked by concomitant ACTH deficiency not receiving replacement therapy, as glucocorticoids are required to eliminate water overload and inhibit antidiuretic hormone secretion.
Other diagnostic markersSometimes additional analytical tests may be necessary to guide a differential diagnosis of hypophysitis, particularly if secondary hypophysitis is suspected. Measurement of systemic inflammation markers, complete blood count and immunoglobulin levels can be helpful for this purpose.
A general assessment should include complete blood count, sedimentation rate, C-reactive protein, calcium, renal function, urinalysis and liver enzymes. A more specific assessment should be based on the clinical characteristics and may include determination of IgG4, angiotensin-converting enzyme, anti-neutrophil antibodies, antinuclear antibodies, other autoantibodies, β2-microglobulin, alpha-fetoprotein, chorionic gonadotropin and a tuberculin test.6 If an infection or cancer is suspected and the initial results are negative, extraction of cerebrospinal fluid for biochemistry, culture, cytology and flow cytometry may be useful.6,13
Imaging testsPituitary magnetic resonance imagingPituitary MRI without and with gadolinium contrast is the imaging test of choice for the diagnosis of hypophysitis. Images must be obtained using instruments larger than 1.5 T with a standardised protocol. High spatial resolution should be achieved with slices of 2−3 mm or thinner and a matrix size of 256 × 512 or above. There are significant advantages to using 3D techniques with ultra-thin slices compared to conventional images.14
The most common findings on MRI in patients with hypophysitis are as follows7,14: 1) moderate increase in gland size; 2) symmetrical suprasellar glandular extension; 3) generally homogeneous contrast uptake; and 4) empty sella turcica as an atrophic response after an inflammatory process. Other less common features are: thickened but not deviated pituitary stalk; absence of bright signal in the posterior pituitary on T1-weighted images; enlargement of the adjacent dura; and thickening of the sphenoid sinus mucosa (Fig. 1).
 and sagittal T1 (B) sequence with paramagnetic contrast in a 59-year-old man with uveal melanoma with liver metastases who developed ipilimumab-induced hypophysitis. Coronal T1 (C) and sagittal T1 (D) pituitary MRI with paramagnetic contrast in a 46-year-old woman with uveal melanoma who developed ipilimumab-induced hypophysitis.")
Ipilimumab-induced hypophysitis. Pituitary MRI with coronal T1 (A) and sagittal T1 (B) sequence with paramagnetic contrast in a 59-year-old man with uveal melanoma with liver metastases who developed ipilimumab-induced hypophysitis. Coronal T1 (C) and sagittal T1 (D) pituitary MRI with paramagnetic contrast in a 46-year-old woman with uveal melanoma who developed ipilimumab-induced hypophysitis.
Pituitary enlargement is usually described as symmetrical in shape, isointense with the brain on T1, and often heterogeneous on T2-weighted images.15 The sellar floor is usually intact in hypophysitis, contrary to what happens in patients with pituitary adenomas. Involvement of the cavernous sinus with ophthalmoplegia has also been described in some patients with hypophysitis.16 MRI abnormalities may not be found in cases of spontaneous resolution of hypophysitis. It is important to note that a normal MRI does not rule out the diagnosis of hypophysitis.
Other imaging testsIn patients with suspected secondary hypophysitis, other imaging tests are indicated to identify possible organ involvement.17 In the diagnosis of sarcoidosis, tuberculosis, connective tissue diseases or malignant tumours, chest or abdomen/pelvis computed tomography is helpful.6
Positron emission tomography/computed tomography with 18F-fluorodeoxyglucose has been described by some authors as helpful in patients with ICPI-induced hypophysitis, with the detection of focal increased uptake in the pituitary gland and its resolution after steroid treatment.18 This imaging test has also been useful to confirm systemic involvement in patients with IgG4-related hypophysitis (IgG4-RH) and so avoid pituitary biopsy.19,20
BiopsyIn clinical practice, the diagnosis of hypophysitis continues to be a diagnosis of exclusion which is made from the clinical, biochemical and imaging data discussed above.21 Hypophysitis should be considered a non-surgical disease and biopsy should therefore be avoided if diagnosis has been possible by other means.
The diagnosis of IgG4-related disease (IgG4-RD) is based on the combination of clinical, serological, radiological and histopathological findings.22,23 The diagnosis of IgG4-RD is essentially established on the histopathology of the specimen, in which dense lymphoplasmacytic infiltration, storiform fibrosis and/or phlebitis obliterans are observed along with IgG4-positive plasma cells (>10–50 per high-power field depending on the organ or a IgG4/IgG ratio >40%).24
The definitive diagnosis of Langerhans cell histiocytosis is infiltration of the pituitary with Langerhans cells containing eosinophils, neutrophils, small lymphocytes and histiocytes.
There are no established criteria for pituitary biopsy in adults. Pituitary biopsy would only be justified in a minority of patients with suspected hypophysitis, to confirm the diagnosis if the clinical and morphological findings were not completely convincing or in cases where the histopathology results are needed to make a decision on treatment (immunosuppression, chemotherapy and/or prolonged corticosteroid therapy).6,7 Pituitary biopsy would be indicated when there are doubts about the differential diagnosis with other diseases with different treatment, such as pituitary adenomas or sellar metastases. Before making a decision on pituitary biopsy, it is important to have multidisciplinary assessment of patients by endocrinologists, neurosurgeons, neuroradiologists and pathologists, when necessary, who are familiar with this rare disease.12
Differential diagnosisHypophysitis has to be differentiated from sellar and parasellar tumours, mainly adenomas, meningiomas, craniopharyngiomas, astrocytomas and germinomas. Metastases, particularly of lung, breast, prostate and kidney cancers, should also be included in the differential diagnosis. Lymphoproliferative processes, such as plasmacytoma or lymphoma, pituitary apoplexy, acute Sheehan's syndrome, and physiological pituitary hyperplasia, such as that seen in untreated severe primary hypothyroidism, must also be differentiated from hypophysitis.6Table 3 shows the differential diagnosis of hypophysitis.
Differential diagnosis of hypophysitis.
| Differential diagnosis | Signs and symptoms | Tests useful for the diagnosis |
|---|---|---|
| Sellar and parasellar tumours: pituitary adenoma, craniopharyngioma, meningioma, astrocytoma and germinoma | Craniopharyngioma: diabetes insipidus, intrasellar or suprasellar cystic images | α-fetoprotein and β-human chorionic gonadotropin levels. Brain imaging studies. Biopsy with or without surgery |
| Germinoma: young male and CDI | ||
| Metastases, particularly breast, lung, prostate and kidney | Sellar mass with or without suprasellar spread. Hypopituitarism and diabetes insipidus. Visual and cranial nerve involvement | Computed tomography |
| FDG PET | ||
| Biopsy | ||
| Lymphoproliferative diseases | Cranial nerve involvement and diabetes insipidus. The pituitary may be the only site involved, but there may be manifestations outside the pituitary | Complete blood count |
| β2-microglobulin | ||
| Lactate dehydrogenase | ||
| FDG PET | ||
| Tissue or bone marrow biopsy | ||
| Pituitary apoplexy | Acute or subacute headache. Visual or cranial nerve defect. Panhypopituitarism without diabetes insipidus. Anticoagulation (risk factor) | MRI with characteristic image |
| Acute Sheehan's syndrome | Acute panhypopituitarism without diabetes insipidus. History of childbirth with heavy bleeding | Postpartum diagnosis |
| Secondary pituitary hypertrophy | Homogeneous pituitary enlargement. Absence of hypopituitarism. Underlying cause: untreated primary hypothyroidism, pregnancy, adolescence | Resolves once the cause disappears |
CDI: central diabetes insipidus; FDG: 18F-fluorodeoxyglucose; PET: positron emission tomography; MRI: magnetic resonance imaging.
Hypophysitis can mimic a non-functioning pituitary adenoma clinically and radiologically. However, unlike the adenoma, most cases of hypophysitis respond well to immunosuppressive therapy.
Various studies have been conducted to evaluate systems, such as machine learning, to differentiate between the two conditions.25 In 2022, Wright et al. proposed a clinical score of 6 points: +2 for diabetes insipidus; +2 for absence of invasion of the cavernous sinus; +1 for infundibular thickening; +1 for absence of visual symptoms. Scores ≥3 supported a diagnosis of hypophysitis (area under the curve 0.96, sensitivity 100% and specificity 75%). The scoring system identified 100% of hypophysitis cases with an estimated false positive rate of 24.7%.26
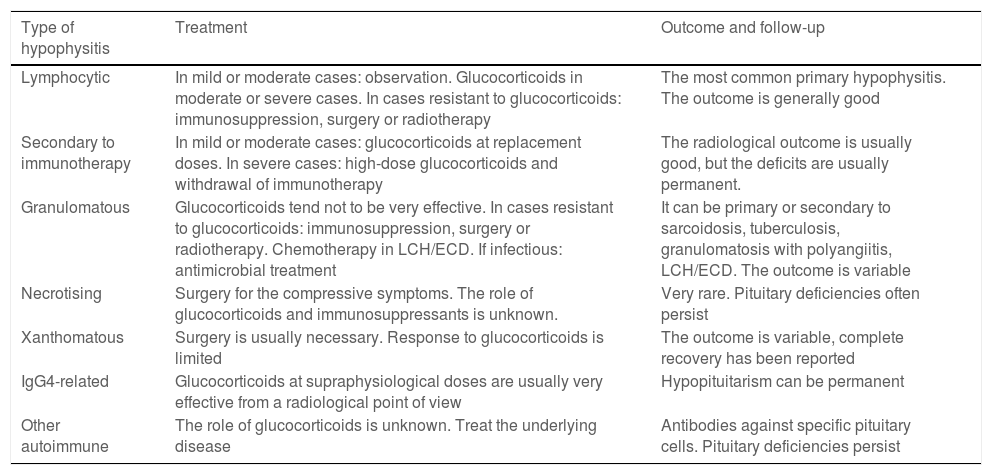
Treatment and follow-upTreatment should be causal and take into account the severity of the patient's clinical condition (Table 4). In patients with a mild degree, replacement therapy can be started and clinical radiological follow-up carried out. In view of their potent anti-inflammatory action, glucocorticoids are considered the mainstay of treatment for primary hypophysitis with compressive symptoms. Recovery of pituitary function does not appear to be linked to treatment with glucocorticoids. The use of glucocorticoids in hypophysitis associated with immunotherapy has to balance the clinical benefit in severe forms of hypophysitis with the need to interrupt the immunotherapy during the treatment. This decision should be made in agreement with Oncology.27
Treatment guide and follow-up of hypophysitis.
| Type of hypophysitis | Treatment | Outcome and follow-up |
|---|---|---|
| Lymphocytic | In mild or moderate cases: observation. Glucocorticoids in moderate or severe cases. In cases resistant to glucocorticoids: immunosuppression, surgery or radiotherapy | The most common primary hypophysitis. The outcome is generally good |
| Secondary to immunotherapy | In mild or moderate cases: glucocorticoids at replacement doses. In severe cases: high-dose glucocorticoids and withdrawal of immunotherapy | The radiological outcome is usually good, but the deficits are usually permanent. |
| Granulomatous | Glucocorticoids tend not to be very effective. In cases resistant to glucocorticoids: immunosuppression, surgery or radiotherapy. Chemotherapy in LCH/ECD. If infectious: antimicrobial treatment | It can be primary or secondary to sarcoidosis, tuberculosis, granulomatosis with polyangiitis, LCH/ECD. The outcome is variable |
| Necrotising | Surgery for the compressive symptoms. The role of glucocorticoids and immunosuppressants is unknown. | Very rare. Pituitary deficiencies often persist |
| Xanthomatous | Surgery is usually necessary. Response to glucocorticoids is limited | The outcome is variable, complete recovery has been reported |
| IgG4-related | Glucocorticoids at supraphysiological doses are usually very effective from a radiological point of view | Hypopituitarism can be permanent |
| Other autoimmune | The role of glucocorticoids is unknown. Treat the underlying disease | Antibodies against specific pituitary cells. Pituitary deficiencies persist |
ECD: Erdheim-Chester disease; LCH: Langerhans cell histiocytosis.
In patients with primary hypophysitis managed exclusively by observation, Honegger et al.28 found that 46% of them showed radiological improvement and a third, hormonal recovery. In the chronic phase, when irreversible changes have occurred, anti-inflammatory treatment is unlikely to affect hormone function or radiological changes.29 There have been no randomised, controlled clinical trials that demonstrate the benefits of treatment with glucocorticoids compared with no therapeutic intervention on the recovery of pituitary function and mass effect. As glucocorticoids have significant side effects, the risks and benefits have to be carefully weighed when deciding on treatment for mild cases of primary hypophysitis. Clinical data should guide the decision to manage the patient by surveillance only. Observation may be appropriate in patients with mild to moderate headache, mild pituitary dysfunction, no optic pathway mass effect, and probable lymphocytic hypophysitis.28,30 Clinical surveillance can be performed with MRI every 3–6 months, with lengthening of the interval if progress is good. However, if the lesions cause a significant mass effect, glucocorticoid treatment should be given or biopsy performed, or both.10 During observation, pituitary function should be assessed periodically. Additionally, patients should be monitored for signs of involvement of other organs which might point to sarcoidosis, Langerhans cell histiocytosis, IgG4-RD or other systemic diseases, which can develop later.31,32
The response of primary hypophysitis to glucocorticoids varies greatly.28,30,33–40 The response in terms of reduction in tumour size can be close to 75%; the rate of hormonal improvement is usually lower.28,30,33,34,36–40 Early treatment with glucocorticoids has been found to improve hormonal recovery.40 In a recent meta-analysis and systematic review on the management of autoimmune lymphocytic hypophysitis, Donegan et al.41 found that treatment with high doses of glucocorticoids was associated with a greater likelihood of hormonal recovery and a decrease in lesions visible on MRI when compared with observation. However, glucocorticoid therapy was associated with a greater likelihood of needing additional treatment after it was reduced or discontinued and careful monitoring is therefore necessary under these circumstances.
The optimal dose and duration of treatment with high doses of glucocorticoids have yet to be clearly established.41 A very wide range of starting doses have been used for the treatment of primary hypophysitis, from 20 mg/day of prednisone to 1 g boluses of methylprednisolone. The exact dose, duration, and even indication of glucocorticoid treatment is the subject of debate. No studies have been conducted comparing high doses or boluses with low doses. A prednisone dose of 1 mg/kg/day with a slow gradual withdrawal may be adequate to reduce side effects and the risk of recurrence.35,42
In patients refractory to treatment with glucocorticoids or as an alternative option, immunosuppression with azathioprine, rituximab or methotrexate can be used. The most studied drug is azathioprine and it seems to be more effective in reducing pituitary size than in hormonal improvement.17,33 Similar results have been found with methotrexate and mycophenolate mofetil.17 Rituximab can be used in diseases in which B lymphocytes predominate and in relapsed IgG4-RD.43–45
Surgery can be used when it is necessary to confirm the diagnosis, for decompression of the chiasm and in cases resistant to glucocorticoids. In a large series of primary hypophysitis, patients who had surgery had worse outcomes.30 However, it is likely that the patients who had surgery had more severe disease.
Fractionated radiotherapy and stereotactic surgery have been used successfully in a small number of patients who required multiple treatment.17,28 Radiosurgery is an option for patients with recurrent and refractory lymphocytic hypophysitis; it helps control the compressive effect and enables immunosuppression to be withdrawn.46,47
Also extremely important is that deficiencies of the different hormones of the anterior and posterior pituitary must be adequately treated.11
The treatment of choice for lymphocytic hypophysitis in patients with severe headache and a significant compressive effect is high doses of glucocorticoids; the response is usually good.48
In hypophysitis secondary to immunotherapy, rapid replacement with hydrocortisone is recommended, followed by evaluation of other possible hormone deficiencies of the anterior pituitary and consideration of replacement therapy. It may be appropriate to delay immunotherapy during the acute stage, but stopping treatment does not appear to improve pituitary function and may aggravate the cancer.49–52 High doses of glucocorticoids should be used when there are severe compressive symptoms, loss of vision, or acute adrenal insufficiency. Adrenal function rarely recovers, but thyroid and gonadal function may recover and should be assessed periodically. There have been proposals for initial hormone monitoring before the start of immunotherapy and follow-up of hormone levels during the treatment, with a common algorithm for all endocrine immune-mediated adverse effects, which could help oncologists in making an early diagnosis of ICPI-induced hypophysitis.53,54
The treatment of choice for granulomatous hypophysitis is glucocorticoids; immunosuppressants such as cyclophosphamide may also be used.55,56 Pituitary tuberculosis is treated with a combination of antituberculosis drugs.57 All other infectious bacterial hypophysitis is treated with antimicrobial therapy.58–60
The classification of histiocytosis and Erdheim-Chester disease as infiltrative diseases or true cancers is the subject of debate.6,61 However, there is agreement on treatment, which includes surgery, glucocorticoids, immunosuppressants, chemotherapy, radiotherapy and targeted therapies (BRAF inhibitors and MEK inhibitors).62
In storage disorders such as haemochromatosis, it has been reported that gonadal function can improve or normalise if iron depletion therapy is instituted early,63 and treatment with copper chelators can lead to hormonal recovery in Wilson's disease.64
IgG4-RH responds very well to pharmacological doses of glucocorticoids and this also prevents fibrosis.65 Doses of 30−40 mg of prednisone per day are usually administered for one to two months and then gradually decreased over two to six months.48 Recurrences are rare and rituximab is a good alternative in these situations.45,66
In the rare autoimmune paraneoplastic hypophysitis,67,68 hypopituitarism is permanent and treatment has to be directed at the cancer, and pituitary hormone replacement is required.
ConclusionsIn conclusion, hypophysitis is a rare disorder which, clinically, can manifest in different ways and be accompanied by different pituitary hormone deficiencies. Histological examination of hypophysitis is the only test that provides a definite diagnosis of the different inflammatory lesions, which include lymphocytic, granulomatous, IgG4-RD, xanthomatous and necrotising hypophysitis. However, most patients do not have a pituitary biopsy and can be managed without histological demonstration, based on clinical judgement made from the history, examination, pituitary function tests and imaging tests, and the response to treatment.
The finding of hypophysitis has become more common in clinical practice in recent times due to the increasing use of ICPI in cancer patients. Both oncologists and endocrinologists should therefore suspect hypophysitis in patients treated with immunotherapy who develop signs or symptoms compatible with pituitary hormone deficiency or who have symptoms of local compression.
