








El riesgo de padecer tumores gonadales de células germinales (TCG) se encuentra incrementado en algunos pacientes con desarrollo sexual diferente (DSD), fundamentalmente en aquellos que presentan material de cromosoma Y. Dicho riesgo, sin embargo, varía considerablemente en función de multitud de factores que dificultan enormemente la decisión de una gonadectomía profiláctica. Con el fin de hacer recomendaciones fundamentadas sobre la conveniencia de este procedimiento en los casos donde existe potencial de malignización, esta guía de consenso evalúa la última evidencia clínica existente, en general escasa, y actualiza los conocimientos en este terreno.
The risk of suffering from gonadal germ cell tumors (GCT) is increased in some patients with different sexual development (DSD), mainly in those with Y chromosome material. This risk, however, varies considerably depending on a multitude of factors that make the decision for prophylactic gonadectomy extremely difficult. In order to make informed recommendations on the convenience of this procedure in cases where there is potential for malignancy, this consensus guide evaluates the latest clinical evidence, which is generally low, and updates the existing knowledge in this field.
La declaración de consenso de las anomalías de la diferenciación sexual del año 2006 estableció el término DSD, del inglés disorders of sex development (anomalías de la diferenciación sexual o, ahora más usado, desarrollo sexual diferente [DSD]), y reorganizó los diagnósticos en tres grupos amplios divididos por sexo cromosómico y, en segundo lugar, por etiología hormonal y genética1.
Las entidades incluidas dentro de los DSD presentan etiologías y expresiones fenotípicas ampliamente variables, si bien, solo un grupo determinado de ellas, especialmente las que presentan material de cromosoma Y, tienen predisposición a desarrollar neoplasias. Estas son, fundamentalmente, los tumores gonadales de células germinales (TCG) (≃14,9% de la totalidad de entidades DSD; 0,8 a 40% según edad y diagnóstico) y, con mucha menor prevalencia (0,9%) y solamente en algunos subtipos de DSD, los tumores del estroma gonadal y los leiomiomas/hamartomas de músculo liso2,3.
Con los avances en la tecnología de diagnóstico molecular se está ampliando de forma considerable el número de genes causantes de DSD, hecho que está permitiendo una taxonomía más precisa. Sin embargo, determinar el riesgo personalizado de neoplasia gonadal (fundamentalmente TCG) en una persona con DSD sigue siendo una tarea desalentadora si tenemos en cuenta que éste varía considerablemente en función de multitud de otros factores no genéticos.
Con el fin de hacer recomendaciones fundamentadas sobre la conveniencia de gonadectomía profiláctica en casos en los que el potencial de malignización no sea desdeñable, este artículo actualiza los conocimientos en este aspecto y evalúa la última evidencia clínica existente, en general escasa, sobre dicho potencial en cada uno de los grupos o entidades incluidos dentro de los DSD.
Todo lo que se expondrá a continuación hará referencia exclusivamente al riesgo de TCG, ya que constituyen las estirpes malignas más frecuentes que pueden asentar en gónadas de pacientes DSD, y en las que los estudios histopatológicos pueden predecir el riesgo de padecerlos.
Ontogenia de las células germinalesEl desarrollo de las células germinales es un proceso muy complejo que está estrictamente organizado en el tiempo y en el espacio sobre una base genética no del todo bien conocida. Tiene su comienzo en la semana 2 a partir de células somáticas que, a través de un proceso de especificación impulsado por BLIMP1 y SOX17, se transforman en células germinales primordiales (CGP). Guiadas por la señalización de KIT/KITLG, tiene lugar la migración de las mismas en grupos a lo largo de la línea media desde el epiblasto proximal hasta el ribete gonadal, proceso que transcurre entre las semanas 5 y 6, y momento en que pasan a denominarse gonocitos (testículo) u oogonias (ovario), todavía indistinguibles morfológicamente de las CGP. Durante este periodo tiene lugar una reprogramación epigenética que les confiere la capacidad de transferir la característica de pluripotencialidad a la siguiente generación, fenómeno que es debido, entre otros factores, a genes de origen parental sometidos a impronta genómica que suponen la expresión, entre otros, de los marcadores PLAP (fosfatasa alcalina placentaria), c-KIT y OCT3/4 (factor de transcripción 3/4 de unión al octámero)4–6.
Conformada la gónada indiferenciada (ribete gonadal y células germinales), la puesta en marcha de una vía de señalización hacia la 7.ª semana determinará la diferenciación de la misma como testículo u ovario. En concreto, los genes SRY y SOX9 serán los principales promotores de la diferenciación testicular, trabajando a través de múltiples factores de transcripción como NR5A1 y ZFPM2 para la formación de las células de Sertoli, mientras que el desarrollo del ovario requerirá de la ausencia de la expresión de SRY, a la vez que la puesta en marcha de vías de señalización de WNT4/β-catenina, FOXL2 y RSPO1, dando lugar a la diferenciación de las células estromales en células de la granulosa. En el testículo, debido a la interacción con las células pre-Sertoli, los gonocitos se diferenciarán en pre-espermatogonias que, como parte de su proceso de maduración, se desplazarán del centro a la lámina basal del tubo seminífero, perdiendo la expresión de los marcadores embrionarios OCT3/4 y c-KIT para, posteriormente, madurar a espermatogonias, adquiriendo expresión de DDX4 y TSPY. Con todo ello, el fin de la diferenciación testicular tendrá lugar en la semana 9, cuando las células de Leydig ya sean capaces de producir testosterona (T) y el factor insulinoide 3 (INSL3), y las células de Sertoli hormona antimülleriana (AMH), mientras que, en el caso del ovario, su diferenciación completa terminará hacia la semana 11 con las células de la teca y la granulosa.
En último lugar, será la presencia de hormonas testiculares (T y AMH) la que determinará la diferenciación genital (interna y externa) masculina y, su ausencia, la femenina5,7.
Patogénesis de las lesiones precursoras de tumor de células germinales en el desarrollo sexual diferentePor definición, las células germinales primordiales son las células más pluripotentes del organismo tras la embriogénesis, en las que la hipometilación y expresión génica son similares a las de las células madre embrionarias. En la mayoría de los pacientes con DSD las células germinales desaparecen conforme el organismo va creciendo mediante un proceso de apoptosis. Algunas pueden persistir en un estado de inmadurez y, en general, se considera que éstas son las células susceptibles de transformación maligna, pasando a ser una lesión premaligna o precursora de TCG.
Los pacientes DSD que corren riesgo de TCG son aquellos que, en presencia de células germinales, contienen material de cromosoma Y en su cariotipo gonadal, situación que sucede con independencia de que exista, o no, algún grado de diferenciación testicular. Este contenido de material Y incluye a determinados genes implicados en la diferenciación gonadal masculina que se encuentran en la región alrededor del centrómero del cromosoma Y (región GBY): TSPY (gen que codifica la «proteína específica del testículo en el cromosoma Y», localizado en Yp11.2) y otros genes candidatos entre los que destacan SRY y DYZ38.
Partiendo de estas premisas, el problema parece residir en que, en ausencia de células de Sertoli debidamente desarrolladas, las células germinales primordiales que migran a la gónada en desarrollo retienen su fenotipo fetal. En estas condiciones de inmadurez biológica, las células germinales pueden malignizarse por acción de los genes mencionados de la región GBY cuya expresión aumentada promueve la proliferación celular, así como una expresión prolongada de OCT3/42,4,6–12. Otro requisito indispensable es que cualquier alteración que pueda suceder a lo largo del desarrollo gonadal tenga un impacto suficiente en la ubicación o entorno de estas células germinales, pero sin llegar a afectar a su supervivencia; en caso contrario, el riesgo de TCG se considerará nulo si dicho entorno es lo suficientemente hostil para inducir su muerte4,12.
Puede concluirse, pues, que el riesgo oncogénico de estas células germinales en presencia de material Y gonadal depende más de las células de sostén (Sertoli) y del microambiente en que se encuentren las propias células germinales; o, lo que es lo mismo, que está relacionado con el grado de diferenciación gonadal, siendo más alto cuanto mayor sea la inmadurez de la gónada.
Con todo ello, se ha estimado que el riesgo de que tales alteraciones se mantengan postnatalmente y terminen en una lesión precursora de TCG infiltrante (con o sin evolución ulterior a este último) es del 14,9% de media en pacientes DSD con material Y. Esto sucede, fundamentalmente, en aquellos pacientes en los que el defecto genético subyacente conduce a un bloqueo precoz del desarrollo gonadal (v.g., mutaciones o deleciones de SRY o WT1). Este riesgo, aunque algo menor, también está presente en el mosaicismo 45X0/46XY (cariotipo gonadal), mientras que resulta significativamente inferior cuando se trata de un DSD que no interfiere con el desarrollo gonadal normal, sino que afecta a la maduración de las células germinales (por ejemplo, las alteraciones en la acción androgénica)2,13.
A estos factores de riesgo de tipo intrínseco deben añadirse otros como el posible papel de las gonadotropinas (especialmente la hormona estimulante del folículo [FSH]), de los estrógenos y, sobre todo, de los andrógenos, lo cual explica el mayor riesgo de desarrollo oncogénico en dos momentos concretos de la vida, la minipubertad y, fundamentalmente, la pubertad/juventud (reinicio de mitosis previamente detenida). También son factores de riesgo la edad del paciente (a mayor edad, mayor riesgo) y la localización gonadal (mayor riesgo en gónadas intraabdominales)2,7,14.
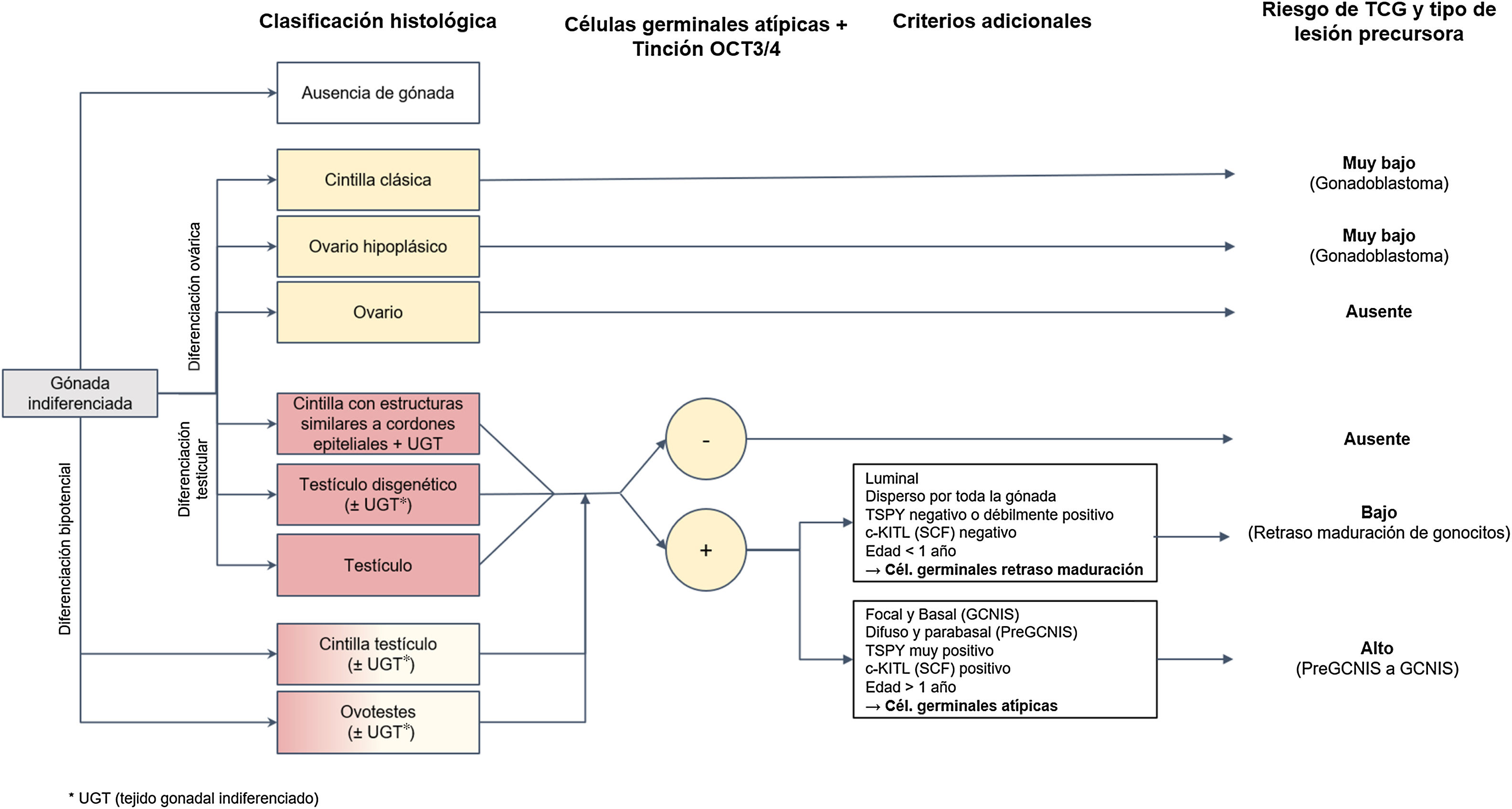
Diagnóstico histopatológico de las lesiones precursoras en pacientes con desarrollo sexual diferente y evolución del riesgo de tumor de células germinales a partir de éstasEl estudio histológico de las gónadas en pacientes DSD permite una clasificación del grado de desarrollo y madurez de las mismas que las agrupan en: (1) cintilla clásica y ovario hipoplásico en gónadas con diferenciación ovárica, (2) cintilla con cordones epiteliales, testículo disgenético y testículo estructuralmente normal en gónadas con diferenciación progresiva hacia testículo, y finalmente, (3) cintilla-testículo y ovotestis en gónadas con diferenciación bipotencial (fig. 1)15. Son este grado de desarrollo, en combinación con alguna de las circunstancias mencionadas, los factores que pueden contribuir en determinados pacientes DSD a que sus gónadas desarrollen lesiones precursoras (menos frecuente en gónadas completamente desarrolladas como testículo, ovario u ovoteste) y que, en última instancia, puedan progresar tarde o temprano a un TCG infiltrante. Estas lesiones precursoras suelen denominarse genéricamente como GCNIS/GB (de Neoplasia in situ de células germinales/Gonadoblastoma) y su diagnóstico está basado en ciertos hallazgos histopatológicos (morfología) combinados, para los casos de DSD con presencia de tejido testicular, con la expresión de determinados marcadores inmunohistoquímicos de pluripotencialidad.
Marcadores de pluripotencialidad en el diagnóstico de lesión precursora de origen testicular: retraso madurativo versus lesión precursora de tumor de células germinales
Como se ha referido, las células germinales en fases iniciales de su ontogénesis expresan determinados marcadores inmunohistoquímicos como OCT3/4 codificado por POU5F1, el Stem Cell Factor (SCF), también conocido como c-KIT ligando (codificado por KITLG) y la fosfatasa alcalina placentaria (PLAP). Tras el nacimiento, aquellas células que mantienen una «maduración retrasada», fundamentalmente las de origen testicular, tienden a conservar la positividad para estos marcadores de pluripotencialidad hasta que entran en apoptosis. Esto explica la no conveniencia de utilizarlos de forma exclusiva en el diagnóstico histológico de lesión precursora de TCG de origen testicular, al menos en los primeros 6-12 meses de vida, y evitar así el sobrediagnóstico durante el periodo postnatal. Para ello es crucial tener en cuenta otros criterios histológicos diferenciadores propios de tales lesiones como son la distribución de las células OCT3/4 positivas en la gónada, la localización de estas células germinales en el túbulo seminífero, la presencia de atipia celular y la expresión del KITLG. En concreto, las células germinales con retraso en la maduración no expresan este último marcador (c-KITLG o SCF), tienden a localizarse en la porción central y suprabasal de los túbulos seminíferos y se distribuyen difusamente por la gónada, mientras que en las lesiones precursoras de TCG (neoplasia in situ de células germinales o GCNIS, como se verá más adelante), sus células expresarán tal factor, tendrán atipia característica, se situarán en situación basal dentro de los túbulos seminíferos en el denominado nicho de la espermatogonia, y tendrán una distribución parcheada5,13.
Precisamente, las gónadas disgenéticas que incluyen parénquima testicular (testículos disgenéticos, cintilla-testículo, cintillas con cordones epiteliales y ovotestes) poseen una mayor incidencia de retraso en la maduración de las células germinales, persistiendo excepcionalmente más allá del primer año de vida5,13,16. Por ello, esta distinción basada no exclusivamente en el marcador OCT3/4 resulta de capital importancia para evitar identificar erróneamente como lesiones precursoras a células inmaduras en gónadas disgenéticas que antaño explicaban frecuencias incorrectas de riesgo de malignización y, por tanto, de evolución a TCG6,7,10,13,17.
En el caso de las gónadas disgenéticas con material Y cromosómico pero que no contienen tejido testicular (cintilla clásica y cintilla con cordones epiteliales), la morfología suele ser suficiente para el diagnóstico de lesión precursora, no siendo necesario el empleo de técnicas inmunohistoquímicas; además, no se plantearía el problema del retraso en la maduración que se ha mencionado.
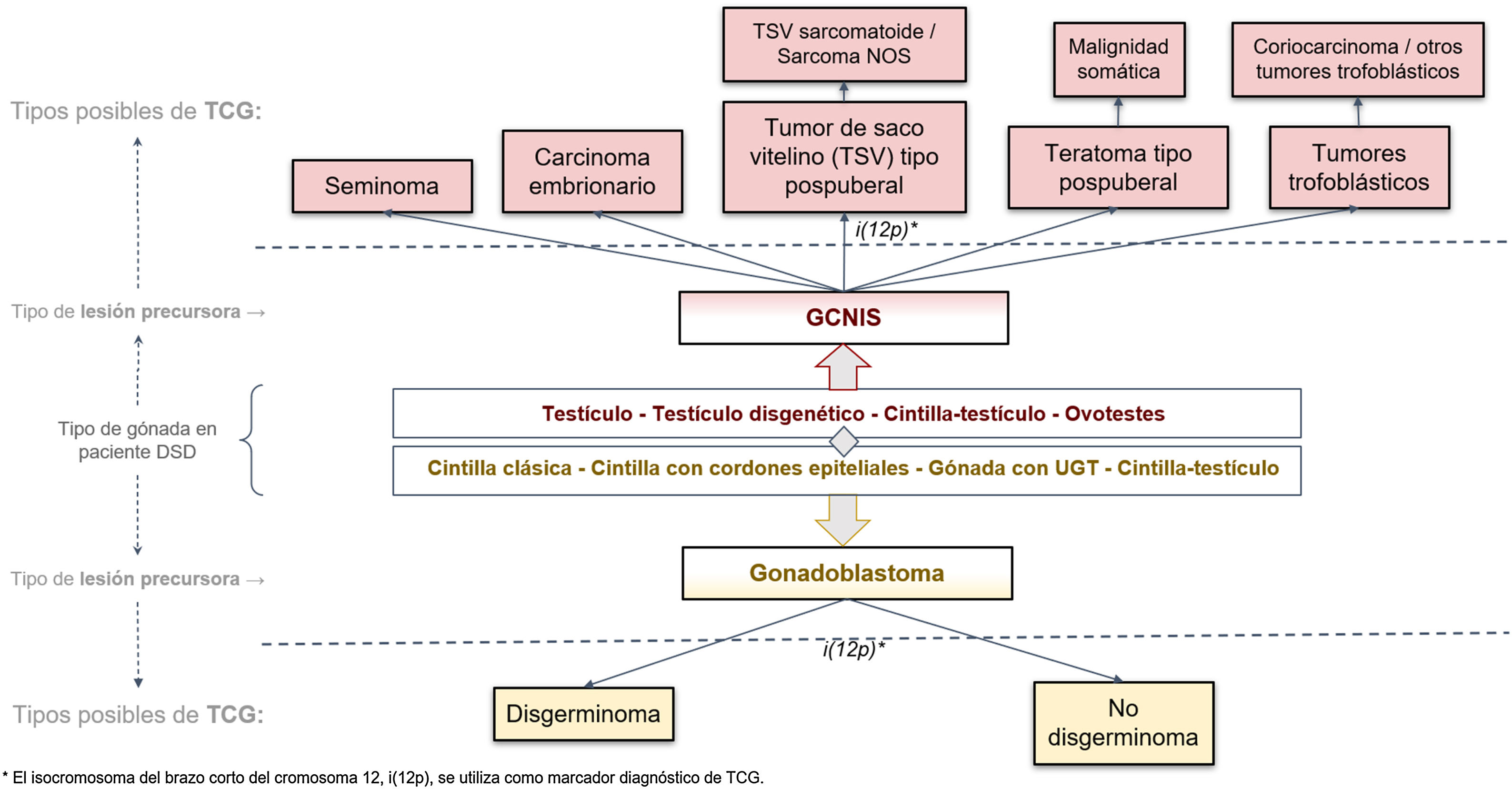
Clasificación histológica de las lesiones precursoras neoplasia in situ de células germinales/gonadoblastoma (GCNIS/GB) y el riesgo hacia un tumor de células germinalesA partir del estudio histopatológico de la pieza de biopsia gonadal y, si procede, de los marcadores inmunohistoquímicos referidos, podrá establecerse el diagnóstico de lesión precursora de TCG (fig. 1). Estas lesiones, denominadas genéricamente GCNIS/GB, se subdividen en 2 tipos según las células de soporte (células de Sertoli o células de la granulosa)4,5,7,9–11,13,18–22:
Gonadoblastoma (GB): habitualmente se produce en pacientes DSD que presentan cintillas u ovario hipoplásico; excepcionalmente aparece en tejido testicular. Son pacientes que presentan cariotipo XY (mosaico o no) pero no expresan el gen SRY, lo que explica que, en muchos casos, sean fenotípicamente mujeres. Su desarrollo está relacionado específicamente con la región GBY, que incluye, entre otros, al mencionado gen TSPY. Así, pacientes 45X0/46XY o mujeres con síndrome de Turner en presencia de material Y tienen mayor riesgo de GB. Sus células de soporte se consideran células de la granulosa (FOXL2 positivo) y se clasifica como un tumor mixto de células germinales y del estroma gonadal. El diagnóstico es principalmente morfológico y no suele requerirse inmunohistoquímica para ello. Consiste en nidos circunscritos formados por una mezcla de células germinales en diferente grado de maduración desde muy similares a las de GCNIS (positivas para OCT3/4) hasta semejantes a espermatogonias y células de los cordones sexuales, en ocasiones rodeadas por depósitos hialinos de material de membrana basal compuestos por laminina.
La evolución de un GB a TCG infiltrante es imposible de predecir. La incidencia real de esta evolución no se conoce dada la tendencia en décadas previas a realizar gonadectomía profiláctica en estas pacientes, si bien, se ha descrito que puede estar presente al nacer o desarrollarse más adelante en la vida. Algunas series establecen que solo el 50% se convertirá en tumor maligno infiltrante a lo largo de la vida, principalmente como disgerminoma9,23.
Neoplasia in situ de células germinales (GCNIS), según denominación de la última clasificación de la WHO 201624; en la literatura médica previa se han utilizado otros sinónimos como CIS (Carcinoma In Situ), incorrecto pues no son células epiteliales, o IGCNU (Intratubular Germ Cell Neoplasia, Unclassified type), equívoco en cuanto a la significación de inclasificable. Esta lesión es considerada lesión precursora de TCG infiltrante de origen testicular que se localiza en los túbulos seminíferos y que, por lo tanto, tiene lugar en pacientes XY, más o menos virilizados, por la presencia del gen SRY. Las células de soporte son células de Sertoli (positivas para SOX9). Las células de GCNIS son grandes y atípicas, semejantes morfológicamente a un gonocito/célula germinal primordial, situadas basalmente en el tubo seminífero, con amplio citoplasma claro y núcleo angulado hipercromático con nucléolo prominente. Para su diagnóstico, además de esta morfología, pueden ser necesarios marcadores inmunohistoquímicos (OCT3/4, c-KIT y PLAP).
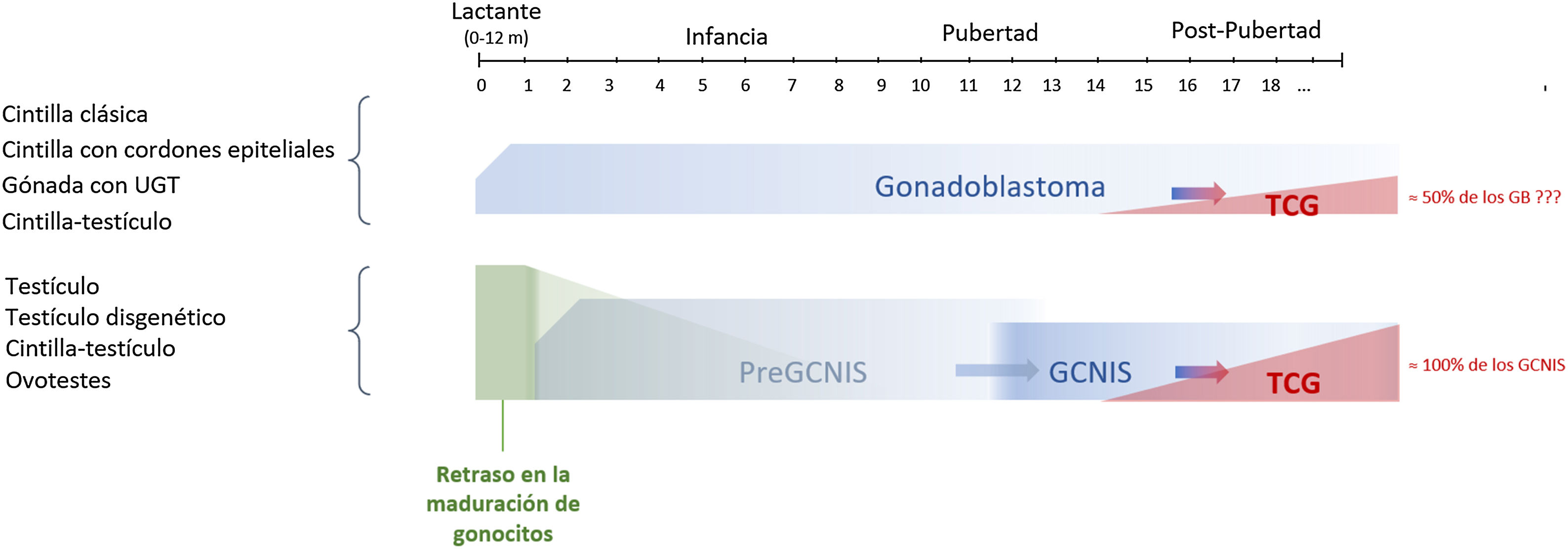
La posibilidad de desarrollo de GCNIS está ya presente al nacimiento por tratarse de un defecto fetal de la maduración de los gonocitos, y casi el 100% está predestinado (algunos autores lo sitúan en el 70%) a desarrollarse como un tumor maligno (TCG) con una edad media de aparición entre los 14 y 44 años de edad (más frecuente entre los 20 y 35 años)9,10,13,23.
Recientemente se ha propuesto el término pre-GCNIS o GCNIS infantil como una lesión intermedia entre el retraso de maduración y el GCNIS que, aunque se sugiere como precursora de esta última, no siempre evoluciona a ella25.
Cuando una de estas lesiones precursoras (GCNIS o GB) progresa hacia un tumor invasivo, lo hace solo hacia estirpes TCG de tipo II (fig. 2). Según la última clasificación WHO 201623, estas estirpes incluyen, en el testículo con GCNIS, al seminoma y los tumores no seminomatosos (carcinoma embrionario, tumor del saco vitelino de tipo pospuberal, teratoma de tipo pospuberal y tumores trofoblásticos de los cuales el más frecuente es el coriocarcinoma), y, en las gónadas disgenéticas tipo cintilla u ovario con GB, al disgerminoma (equivalente al seminoma testicular)9,13,23.

Clasificación histológica de los TCG según la clasificación WHO 201624 aplicada a gónadas disgenéticas.
DSD: desarrollo sexual diferente; GCNIS: neoplasia in situ de células germinales; TCG: tumores gonadales de células germinales; UTG: tejido gonadal indiferenciado.
La cronología de esta evolución desde un tejido gonadal inicial hasta un TCG, pasando previamente por lesiones precursoras GCNIS/GB, se muestra en la figura 3.
El binomio riesgo/beneficio de la gonadectomía profiláctica y la toma de decisiones en pacientes con desarrollo sexual diferente con riesgo de tumor de células germinales y neoplásicos (TCG) en gónadas DSD con material Y. GCNIS: neoplasia in situ de células germinales; TCG: tumores gonadales de células germinales; UTG: tejido gonadal indiferenciado.")
La gonadectomía profiláctica solamente es planteable en pacientes DSD con riesgo para TCG. El riesgo de padecer otras estirpes neoplásicas como los tumores del estroma gonadal (referido a los hamartomas de Sertoli-Leydig, adenomas de células de Sertoli y los leiomiomas/hamartomas de músculo liso, característicos del síndrome de insensibilidad completa a andrógenos), no justifica per se la gonadectomía profiláctica por su baja frecuencia y su carácter casi siempre benigno.
Clásicamente, en la toma de decisiones para la realización de la gonadectomía en los pacientes con verdadero riesgo de TCG se han tenido en cuenta 3 factores: el diagnóstico subyacente, el sexo de crianza y la edad de presentación, añadiéndose recientemente los hallazgos moleculares, histopatológicos e inmunohistoquímicos3,8,19. Pese a los últimos avances en estos campos, los riesgos mencionados de malignización (fundamentalmente en los GB) son meras especulaciones teóricas, desconociéndose la historia natural real de los mismos, tanto en tasas de malignización como en los tiempos de desarrollarlos4,5,7,17. Estas limitaciones obligan a tener en cuenta que una gonadectomía implica infertilidad, la necesidad de suplementación hormonal y la parte subjetiva referida por algunos pacientes de una peor adquisición de caracteres sexuales secundarios, así como un empeoramiento de la calidad de vida pese a una adecuada terapia sustitutiva. Por otro lado, de no optarse por ella, pese a que la tasa de supervivencia de un TCG maligno es elevada (95% a los 5 años), las terapias quimioterápicas de estos entrañan efectos secundarios de por vida como el riesgo de síndrome metabólico y cardiovascular5.
Dicho esto, a día de hoy, en los pacientes DSD con actividad gonadal (habitualmente testículos) hay una tendencia creciente a posponer, e incluso evitar, la gonadectomía, equilibrando cuidadosamente el riesgo de desarrollar un TCG por un lado, con el mantenimiento de la función endocrina y del potencial de fertilidad por el otro. En este empeño, la biopsia gonadal sigue siendo el estándar de oro para la evaluación de este riesgo en determinados DSD (tabla 1) y, como pauta general de actuación, se recomienda que la muestra de biopsia sea de un tamaño de 3×3×2mm al objeto de disponer de una cantidad suficiente de tejido para el examen anatomopatológico, a la par que requiere de patólogos experimentados en esta área para garantizar una interpretación adecuada de las lesiones. No podemos olvidar, no obstante, que la biopsia no representa necesariamente toda la gónada y que puede escaparse el diagnóstico de una lesión precursora por lo que, en las gónadas con aspecto macroscópico heterogéneo, como por ejemplo los ovotestes, debe procederse a la toma de muestras de diferentes áreas. Además, algunos autores recomiendan que la práctica de la biopsia se combine con otros procedimientos como la orquidopexia o la extracción de esperma en el caso de DSD testicular7,8,10.
Indicaciones para la biopsia gonadal en pacientes DSD*
| • En prepubertad, a partir del año de edad (sin haber una clara recomendación, se propone entre 1 y 2 años): |
| ∘ Pacientes DSD 46XY con sospecha clínico-hormonal de disgenesia gonadal |
| ∘ Pacientes DSD 45X0/46XY |
| ∘ Pacientes DSD 46XX con hipervirilización de origen gonadal (46XX ovotesticular y 46XX testicular) |
| • En pubertad o pospubertad, a valorar en las siguientes situaciones: |
| ∘ Los pacientes DSD previamente mencionados tras primera biopsia (repetir en pubertad o tres finalizarse ésta) |
| ∘ Pacientes DSD 46XX/46XY (en pubertad a la par que la exéresis de parte del tejido gonadal no deseado, o en pospubertad) |
| ∘ A valorar si no hay deseo de gonadectomía en pacientes DSD 46XY por anomalías del receptor de andrógenos (en pospubertad) |
El objetivo de la biopsia es el estudio histopatológico para clasificar la gónada y, en caso de tejido testicular y según morfología, valorar estudio inmunohistoquímico (retraso en la maduración de gonocitos [0-12 meses de edad] versus preGCNIS [a partir del año de edad y antes de la pubertad] y GCNIS [habitualmente a partir de la pubertad]).
De igual manera, a excepción del síndrome de Turner 45X0/46XX o 45X0 sin material de cromosoma Y (demostrado mediante cariotipo convencional, FISH y/u otras técnicas moleculares), si se sospecha que las gónadas sean cintillas cuando son rudimentarias, éstas deben ser siempre examinadas histológicamente para descartar la presencia de tejido gonadal indiferenciado o UGT (lesión precursora de GB), que tiene una alta probabilidad de desarrollar un TCG con el tiempo. Por este motivo, y debido a que desde un punto de vista funcional estos tejidos no tienen ningún tipo de actividad hormonal, debería procederse siempre a su exéresis10.
Según el resultado del examen histológico, y antes de decidir la realización de la gonadectomía (tabla 2), se debe hacer una estratificación del riesgo de desarrollar un TCG y considerar otros factores, algunos ya mencionados, como la dotación cromosómica y el diagnóstico subyacente, la edad del paciente (mayor riesgo a mayor edad), la raza (riesgo incrementado en la caucásica), la situación anatómica de la gónada (mayor en la intraabdominal), el fenotipo genital, la fertilidad potencial, la calidad de la actividad endocrina de la gónada, la posibilidad de efectuar un autoexamen, los riesgos inherentes de la cirugía y los posibles efectos secundarios del tratamiento hormonal sustitutivo3,8,10,14. A estos factores deben añadirse otros de índole psicológica, no menos importantes, como el grado de entendimiento del riesgo y de cooperación por parte del paciente, así como la congruencia con su identidad de género. Todos ellos en conjunto, y sobre la base fundamental de una evaluación del grado de desarrollo de esta identidad, deben tenerse en cuenta a la hora de establecer estrategias de actuación y seguimiento. Es por ello que, desde los inicios del abordaje médico de un DSD y, fundamentalmente durante la adolescencia, deba explorarse la identidad de género de estos pacientes y trabajar sobre ella para que cualquier decisión quirúrgica pueda tomarse con mayores garantías de éxito26. Téngase en cuenta, no obstante, que estudios recientes muestran que la mayoría de los individuos adultos con DSD se identifican con el género asignado al nacer, cercano al de la población general, si bien, cuando no existe tal congruencia, la tasa y el grado de disforia de género que existe se han constatado como muy elevados27.
Criterios que aconsejan la gonadectomía en pacientes DSD
| • Diagnóstico histopatológico de GCNIS*/GB (obligado en TCG): |
| ∘ En caso de GCNIS, se debe informar a los padres que la presencia de dicha lesión en la biopsia es precursora de un TCG infiltrante en prácticamente el 100% de los casos (menos para algunos autores), en un tiempo de aparición variable (de 5 a 10 años) y con posibilidad de evolución a ser metastásico. |
| ∘ En el caso de GB, aunque existe riesgo de TCG, éste se considera menor (se desconoce la cifra exacta). |
| • Diagnóstico histopatológico de tejido gonadal indiferenciado (UGT). Considerado precursor de GB por algunos autores. |
| • Detección sanguínea de material Y mediante cariotipo/FISH o estudio molecular para genes de la región GBY (TSPY, SRY y DYZ3, entre otros) en pacientes con síndrome de Turner. Aunque no se realiza habitualmente, podría plantearse FISH en busca de material de cromosoma Y en la gónada. |
| • A valorar cuando el género sentido es diferente al sexo gonadal o la gónada no es funcionante. |
Cuando el paciente con DSD, o sus padres, desean finalmente retener las gónadas pese a que el genotipo y/o el análisis histopatológico e inmunohistoquímico muestren un potencial de malignización no desdeñable, se les puede ofrecer una estrategia basada en la detección proactiva precoz de lesiones localizadas sugerentes de TCG mediante imagen ecográfica periódica a nivel inguinal o escrotal, y/o mediante la autoexploración si las gónadas son fácilmente palpables (localización escrotal). Si, por el contrario, la localización de las gónadas es intraabdominal, dado que las imágenes se han demostrado insuficientes a este nivel, se han propuesto opciones alternativas como la reubicación de las mismas a nivel de canal inguinal para una mejor visualización. Sean estos, u otros, los motivos para retener las gónadas, se aconseja un seguimiento de las mismas a partir del comienzo de la pubertad según se expone en la tabla 35,8.
Propuesta de seguimiento en pacientes DSD con riesgo de TCG* desde el inicio puberal hasta la gonadectomía (o indefinidamente si hay deseo de preservar gónadas)
| • Autoexploración gonadal de por vida, y palpación periódica por el pediatra/médico de cabecera. |
| • Ecografía gonadal anual a partir de la pubertad (en prepúberes si alto riesgo) en las de localización inguinal o escrotal. Se propone, en las de localización intraabdominal, orquidopexia inguinal o escrotal. |
| • Detección anual de alfa-fetoproteína, beta-hCG y LDH a partir de la pubertad (en prepúberes si alto riesgo) como marcadores de algunos TCG** (tumor del saco vitelino y coriocarcinoma). Alternativa que aún no tiene validez clínica: la detección de microRNA en plasma. |
En este grupo pueden incluirse aquellos pacientes en los que la biopsia no muestra riesgo histopatológico de TCG (GCNIS/GB) pero sobre la que se concluye que podría no ser representativa y donde, además, haya algún factor de riesgo como la presencia de tejido testicular, de material Y o la ubicación intraabdominal de la gónada.
Aumentos de beta-hCG muy importantes (de varios miles) se asocian a la presencia de coriocarcinoma, mientras que aumentos leves del mismo (de cientos a 1-2 mil) se asocian a presencia de células de sincitiotrofoblasto en cualquier otro TCG. Por otro lado, niveles altos de alfa-fetoproteína son propios del tumor de saco vitelino. No hay aumento de tales marcadores en el seminoma (o disgerminoma en el ovario), el carcinoma embrionario ni el teratoma de tipo pospuberal.
Por último, y sea cual fuere la decisión tomada respecto a la gonadectomía, debe informarse al paciente y sus padres sobre las posibilidades de fertilidad. En primer lugar, es preciso analizar el riesgo de transmitir a la descendencia la condición DSD genéticamente determinada y, por el otro, investigar la presencia de células germinales en la biopsia del tejido gonadal, número que, en general, se reduce considerablemente con la edad, llegando a estar ausente o muy disminuido tras la pubertad en la mayoría de estos pacientes. Este dato de celularidad, no obstante, es muy dependiente de la entidad que se padezca, de modo que puede estar ausente o reducida ya desde el nacimiento en una gran proporción de las disgenesias gonadales, o ser normal o casi normal en sus inicios, pero en reducción progresiva y con predominio de formas inmaduras, en pacientes con insensibilidad completa a los andrógenos o en los defectos leves de la síntesis de andrógenos como el déficit de 5-alfa reductasa. En todo caso, aunque las técnicas de preservación de tejido germinal aplicables hoy en día son factibles (criopreservación de tejido precursor en fases tempranas de la vida o, rara vez, de espermatozoides u ovocitos en algunos casos leves de DSD con inicio puberal espontáneo), se requiere de protocolos de investigación muy estrictos para las técnicas de inducción de fertilidad futura de células germinales inmaduras (maduración in vitro), que ya están conseguidas para el tejido germinal ovárico, pero aún no disponibles en la actualidad para las de origen testicular si no es de forma experimental28,29.
Propuesta para la indicación de gonadectomía profiláctica en el desarrollo sexual diferenteA modo de resumen, las recomendaciones actuales de gonadectomía profiláctica están basadas en el tipo de DSD y se detallan en la tabla 4. La irradiación de las gónadas como alternativa se ha propuesto pero la experiencia es escasa8.
Propuesta de consenso para la realización de gonadectomía según tipo de DSD
| Grupo | Entidad | Riesgo | Recomendación de gonadectomía y seguimiento |
|---|---|---|---|
| DSD con anomalías en el cariotipo | 45X0/46XY (Disgenesia gonadal mixta) | Intermedio- Alto para TCG(15-35%) | Se recomienda la gonadectomía, aunque se puede valorar diferir según grado de virilización, identidad de género y deseo genésico (si se constata potencial de fertilidad) 2,8,26,27
|
| 46XY/46XX (quimera ovotesticular: cintilla-testículo u ovotestes) | Bajo o muy bajo para TCG(2,6-3%) | La gonadectomía se indicará en la pubertad, y solo de la gónada discordante (teste u ovario) con el género asignado 5,8,28–31:
| |
| 45X0/46XX(Sd. Turner) | Casi nulo o Intermedio para TCG (GB) según se detecte material de cromosoma Y o no(0-1% y 12-40% respectivamente) | Todo síndrome de Turner, fundamentalmente el 45X0 no mosaico, requiere de la detección de material Y a nivel sanguíneo (cariotipo/FISH seguido de estudio molecular de genes de región GBY como TSPY, SRY y DYZ3, entre otros) 32–37:
| |
| 47XXY(Sd. Klinefelter) | Nulo para TCG | No está indicada la gonadectomía, no requiere biopsia ni tampoco seguimiento específico a nivel gonadal. El riesgo de TCG está descrito exclusivamente a nivel extragonadal. Se ha descrito, de manera excepcional, tumores de células de Leydig 2. | |
| 47XYY | Nulo para TCG | No está indicada la gonadectomía, no requiere biopsia ni tampoco seguimiento específico a nivel gonadal. | |
| DSD XX con hipervirilización | Hipervirilización de origen NO gonadal (hiperplasia suprarrenal congénita, tumores virilizantes, etc.) | Nulo o casi nulo para TCG | En términos generales no está indicada la gonadectomía, no requiere biopsia ni tampoco seguimiento específico a nivel gonadal:
|
| Hipervirilización de origen gonadal (DSD ovotesticular 46XX y DSD testicular 46XX) | En ausencia de TSPY (± SRY), posiblemente riesgo bajo o muy bajo (2,6-3%) para TCG.En presencia de TSPY posiblemente riesgo Intermedio del tejido testicular para TCG. | La biopsia gonadal suele ser necesaria para el diagnóstico definitivo en donde, además, se sugiere la realización de un cariotipo para descartar el mosaicismo gonadal, así como el estudio molecular de material de cromosoma Y para establecer riesgo de TCG (al menos TSPY y SRY).Este grupo de disgenesia gonadal 46XX suele tener una causa genética como la translocación del gen SRY al cromosoma X o a un autosoma (10-15% de los casos de DSD ovotesticular, u 80% de los de DSD testicular), o más ocasionalmente, a la duplicación de SOX9, mutaciones en RSPO1, SOX10, NR5A1, NR2F2, WNT4 o WT1. Pocas veces subyace un mosaicismo gonadal 46XX/46XY en los que el riesgo de TCG parece ser igualmente bajo (véase entidad 46XY/46XX) 5,8,28–31.La gonadectomía se indicará en la pubertad, ysolo de la gónada discordante (teste u ovario) con el género asignado. Véase apartado previo 46XY/46XX:
| |
| DSD XY | Disgenesia testicular, completa y parcial (DGC y DGP)DSD XY ovotesticularDSD XY ovárico | Intermedio - Alto para TCG (12-60%) según alteración molecularBajo para TCG(2,6-3%)No descrito. Posiblemente bajo. | La mayoría de los casos precisan de biopsia gonadal para el diagnóstico definitivo. La decisión de gonadectomía en este amplio subgrupo es complicada ya que el riesgo de TCG varía ampliamente según diversos factores: (1) la presencia del gen TSPY (Yp.11.2) [la detección de SRY sólo indica que existe material del cromosoma Y, pero no es útil como marcador de predisposición tumoral] y de un gen causante (cuando se encuentra, pese a que los riesgos no suelen estar bien establecidos) y, por otro lado, (2) las características de la gónada: posición anatómica (escrotal, inguinal o abdominal) y estadio madurativo (no diferenciada o UGT, ovario, testículo u ovotestes), siendo menor el riesgo tumoral cuanto más madura es la gónada y más baja sea su localización 5,8,12,13,17,40. Para más detalles, se propone lo siguiente:
|
| Anomalías de la síntesis de andrógenos | No bien conocido (variable según entidad) | Aunque el riesgo, en general, parece bajo o muy bajo, determinadas entidades parecen tener riesgo de TCG. La decisión de gonadectomía dependerá, por tanto, del diagnóstico molecular y, también, del género asignado 5,8,46,47:
| |
| Síndrome de insensibilidad a los andrógenos | Formas completas (CAIS) sin actividad residual de receptor AR:Bajo riesgo de TCG#(riesgo variable entre estudios: 1-3% con un riesgo acumulado de un 3,6% a los 25 años, y 33% a los 50 años 48, otros autores estiman un riesgo del 15% pasada la pubertad 49. | Al desconocerse en buena medida el riesgo de malignización en la edad adulta, las decisiones en torno a la gonadectomía en CAIS siguen envueltas en una considerable controversia. En cualquier caso, la recomendación más aceptada actualmente es la gonadectomía en pubertad tardía ya que: (1) el TCG (seminoma) se presenta después de la pubertad, y (2) si se mantienen las gónadas hasta la etapa postpuberal se producirá una feminización puberal espontánea (por aromatización de los andrógenos) sin necesidad de estrógenos exógenos, lográndose una adecuada optimización tanto del desarrollo mamario como de la mineralización ósea. Además, (3) diferir la realización de la gonadectomía permite a la paciente participar en la decisión tras haber sido adecuadamente informada 8,50,51:
| |
| Formas parciales (PAIS)y CAIS con actividad residual del receptor AR:Intermedio - Alto riesgo de TCG(15-20%).Menor, si localización escrotal; estudios recientes reducen este riesgo sustancialmente. | No existen estudios de seguimiento de pacientes con PAIS en los que se difiera la gonadectomía, por lo que la actitud predominante es la gonadectomía bilateral precoz51.
| ||
| Síndrome de persistencia de los conductos müllerianos | Bajo o muy bajo para TCG | No está indicada la gonadectomía ni requiere biopsia, si bien, los raros casos de TCG descritos podrían aconsejar un seguimiento a partir de la pubertad o más tardíamente (véase tabla 3) 3. |
beta-hCG: gonadotropina coriónica; DSD: desarrollo sexual diferente; FSH: hormona folículo estimulante; GBY: región alrededor del centrómero del cromosoma Y; GCNIS/GB: neoplasia in situ de células germinales/gonadoblastoma; GnRH: hormona liberadora de gonadotropina; LDH: lactato deshidrogenasa; TCG: tumores gonadales de células germinales.
