




La neoplasia endocrina múltiple tipo 1 (MEN 1) es una rara enfermedad hereditaria (su prevalencia aproximada es de 2:100.000) autosómica dominante por mutación del gen MEN1, ubicado en el cromosoma 11q13, que codifica la menina (una proteína nuclear reguladora del ciclo celular). Predispone al desarrollo de tumores, sobre todo en las glándulas paratiroides, las células endocrinas enteropancreáticas y la hipófisis anterior, pero también en otras numerosas localizaciones, incluyendo la corteza suprarrenal. Su penetrancia es tan alta (alrededor del 98% de los pacientes afectos desarrollará alguna neoplasia en las primeras 5 décadas de la vida) que está recomendado un seguimiento estricto desde la edad de 5 años. La misma mutación puede producir distintos fenotipos en cada familiar afecto. La aparición de un hiperandrogenismo en un paciente con MEN 1 a cualquier edad obliga a descartar un tumor suprarrenal1.
Presentamos un niño en seguimiento desde los 5 años de edad por ser portador de la mutación p.ala337asp del gen de la menina tras estudio familiar (padre con la misma mutación, intervenido de adenoma de paratiroides a la edad de 30 años). A los 7 años presenta pubarquia incipiente, engrosamiento del pene (30×15mm) sin aumento del tamaño testicular (2mL) e hipercrecimiento (peso 26,3kg, talla 131,7cm [p92]), IMC 15,16kg/m2 (p28), velocidad de crecimiento 8,6cm/año (p>99). La edad ósea estaba adelantada 1,5 años. Se realizó una RMN abdominopélvica que fue informada como normal, y un estudio hormonal con los siguientes resultados: 17-hidroxiprogesterona 55,7nmol/L (normal<5), testosterona total 0,6nmol/L (normal<0,7), dehidroepiandrosterona-sulfato 4,8μmol/L (normal<4,0), androstendiona 8,9nmol/L (normal<4,2), LH 0,1U/L y FSH 0,3U/L; el patrón bioquímico era compatible con hiperplasia suprarrenal congénita. Se inició tratamiento con hidrocortisona 15mg/m2/día repartidos en 3 dosis, comprobándose un descenso de la velocidad de crecimiento a 4,8cm/año (p15), de los niveles de andrógenos (testosterona 0,3nmol/L, dehidroepiandrosterona-sulfato 3,6mmol/L, androstendiona 4,3nmol/L) y de 17-hidroxiprogesterona (12nmol/L). El estudio del gen de la 21-hidroxilasa (gen CYP21A2, localizado en el cromosoma 6p21) demostró 2 mutaciones en heterocigosis compuesta (Ile173Asn/Val282Leu) en el paciente y una en cada uno de sus progenitores: Ile173Asn en el padre y Val282Leu en la madre. Con la confirmación diagnóstica de forma tardía de hiperplasia suprarrenal congénita por déficit de 21-hidroxilasa se ha continuado el tratamiento con hidrocortisona para evitar el avance de la edad ósea, así como el protocolo de despistaje de neoplasias del MEN1.
Entre el 20 y el 70% de los pacientes con MEN 1 presentan alguna lesión suprarrenal a lo largo de su vida, excepcionalmente en la infancia. La mayoría son tumores benignos, unilaterales y no funcionantes, aunque se han descrito algunos productores de cortisol, aldosterona y, más raramente, de andrógenos y feocromocitoma. Los funcionantes requieren tratamiento, así como aquellos que presentan algún riesgo de malignidad: los mayores de 4cm y los que crecen o presentan una imagen atípica o indeterminada. En los no funcionantes más pequeños se tiende a mantener una actitud conservadora, pues en su seguimiento las lesiones no suelen cambiar de aspecto ni aumentar de tamaño2-7.
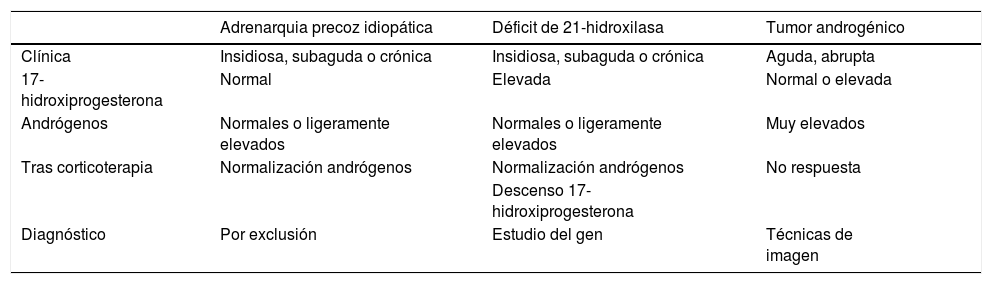
Ante un niño con MEN 1 e hiperandrogenismo se debe plantear el diagnóstico diferencial de la enfermedad tumoral con las entidades propias de la edad, como son la forma tardía de hiperplasia suprarrenal congénita y la adrenarquia precoz idiopática. El perfil hormonal, la respuesta al tratamiento corticoideo, las pruebas de radiología y el estudio genético establecerán el diagnóstico (tabla 1). Descartado el origen tumoral, debemos pensar que se trata de otra enfermedad concomitante. No hemos encontrado previamente descrita en la bibliografía esta asociación de enfermedades. Aunque ambas sean genéticas, las producen genes diferentes y distantes entre sí, por lo que su asociación debe ser totalmente casual.
Diagnóstico diferencial del hiperandrogenismo en la infancia
| Adrenarquia precoz idiopática | Déficit de 21-hidroxilasa | Tumor androgénico | ||
|---|---|---|---|---|
| Clínica | Insidiosa, subaguda o crónica | Insidiosa, subaguda o crónica | Aguda, abrupta | |
| 17-hidroxiprogesterona | Normal | Elevada | Normal o elevada | |
| Andrógenos | Normales o ligeramente elevados | Normales o ligeramente elevados | Muy elevados | |
| Tras corticoterapia | Normalización andrógenos | Normalización andrógenos | No respuesta | |
| Descenso 17-hidroxiprogesterona | ||||
| Diagnóstico | Por exclusión | Estudio del gen | Técnicas de imagen |
