


Kennedy's disease, also known as bulbospinal muscular atrophy, is a rare, X-linked recessive neurodegenerative disorder affecting adult males. It is caused by expansion of an unstable cytosine-adenine-guanine tandem-repeat in exon 1 of the androgen-receptor gene on chromosome Xq11-12, and is characterized by spinal motor neuron progressive degeneration. Endocrinologically, these patients often have the features of hypogonadism associated to the androgen insensitivity syndrome, particularly in partial forms.
We report 4 cases with the typical neurological presentation, consisting of slowly progressing generalized muscle weakness with atrophy and bulbar muscle involvement; these patients also had several endocrine manifestations; the most common non-neurological manifestation was gynecomastia. In all cases reported, molecular analysis showed an abnormal cytosine-adenine-guanine triplet repeat expansion in the androgen receptor gene.
La enfermedad de Kennedy o atrofia muscular espino-bulbar es un trastorno neurodegenerativo raro de herencia recesiva ligada al cromosoma X que afecta a varones en la edad adulta. Está causado por la expansión de secuencia repetida citosina-adenosina-guanina (en el exón 1 del gen del receptor androgénico localizado en el cromosoma Xq11-12, y se caracteriza por la degeneración progresiva de las neuronas motoras espinales. Desde el punto de vista endocrinológico es común encontrar en estos pacientes datos de hipogonadismo englobados en el síndrome de resistencia androgénica, particularmente la forma parcial.
Se describen 4 casos con presentación clínica neurológica típica de la enfermedad, con debilidad muscular generalizada lentamente progresiva con atrofia y afectación de musculatura bulbar; entre las manifestaciones endocrinológicas observadas la ginecomastia fue la más frecuente. El estudio molecular mostró una expansión anormal del triplete citosina-adenosina-guanina en el gen del receptor androgénico en todos los casos.
Kennedy's disease (KD), or spinal and bulbar muscular atrophy, is a progressive neurodegenerative disorder of the motor neuron starting in adult age1 which was first described in 1968 and whose genetic cause was established in 1991.2 The reported prevalence of KD is 3.3/100,000 population in European Caucasian males,3,4 but it is also thought that there is a large proportion of underdiagnosed cases.5
KD is an X-linked recessive disorder and clinically affects males.6 KD has its origin in a dynamic mutation in the androgen receptor (AR) gene located in Xq11-q12, which consists of an abnormal expansion of the cytosine-adenine-guanine (CAG) triplet in exon 1 of the gene.2,3,6–8
Clinically, patients experience progressive weakness in limb muscles and atrophy, fasciculation, tremor, or cramps in the bulbar muscles. Endocrine manifestations consist of hypogonadism resulting from androgen resistance syndrome.3,9–11
The disease is diagnosed by showing the presence of more than 40 CAG triplets in the AR gene.2,3,6
Four cases of KD confirmed by genetic study are reported.
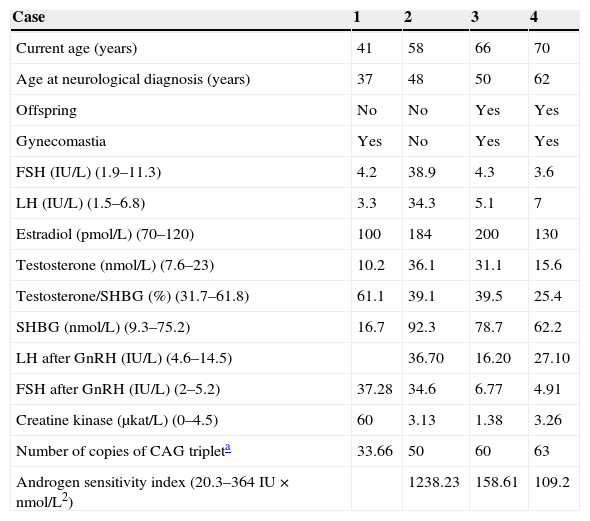
Patients and methodsThe individual clinical, laboratory, and genetic data of four patients are reported and summarized in Table 1.
Clinical and hormonal characteristics and genetic study of the four patients.
| Case | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Current age (years) | 41 | 58 | 66 | 70 |
| Age at neurological diagnosis (years) | 37 | 48 | 50 | 62 |
| Offspring | No | No | Yes | Yes |
| Gynecomastia | Yes | No | Yes | Yes |
| FSH (IU/L) (1.9–11.3) | 4.2 | 38.9 | 4.3 | 3.6 |
| LH (IU/L) (1.5–6.8) | 3.3 | 34.3 | 5.1 | 7 |
| Estradiol (pmol/L) (70–120) | 100 | 184 | 200 | 130 |
| Testosterone (nmol/L) (7.6–23) | 10.2 | 36.1 | 31.1 | 15.6 |
| Testosterone/SHBG (%) (31.7–61.8) | 61.1 | 39.1 | 39.5 | 25.4 |
| SHBG (nmol/L) (9.3–75.2) | 16.7 | 92.3 | 78.7 | 62.2 |
| LH after GnRH (IU/L) (4.6–14.5) | 36.70 | 16.20 | 27.10 | |
| FSH after GnRH (IU/L) (2–5.2) | 37.28 | 34.6 | 6.77 | 4.91 |
| Creatine kinase (μkat/L) (0–4.5) | 60 | 3.13 | 1.38 | 3.26 |
| Number of copies of CAG tripleta | 33.66 | 50 | 60 | 63 |
| Androgen sensitivity index (20.3–364IU×nmol/L2) | 1238.23 | 158.61 | 109.2 |
The normal range of values is given in brackets.
SHBG: sex hormone-binding globulin; CAG triplet: cytosine-adenine-guanine.
FSH, LH, and SHBG were measured using non-competitive solid-phase chemiluminescence enzyme immunoassay with an incubation temperature of 37°C, an incubation time of 30min, and non-linear calibration with two calibrators (Immulite 2000 analyzer, Siemens). The chemiluminescence measured is directly proportional to the amount of hormone present in the sample. The reference values for FSH and LH are 1.9–11.3IU/L and 1.5–6.8IU/L, respectively; normal SHBG values range from 9.3 to 75.2nmol/L.
Estradiol and total testosterone were measured using competitive solid-phase chemiluminescence enzyme immunoassay with an incubation temperature of 37°C, and incubation time of 60min, and non-linear calibration using two calibrators. The chemiluminescence measured is inversely proportional to the amount of hormone present in the sample. Reference values are 7.6–23nmol/L for testosterone and 70–120pmol/L for estradiol. The normal range for the testosterone/SHBG ratio is 31.7–61.8%.
The product of absolute serum testosterone level and LH level is known as the androgen sensitivity index (ASI).
Creatine kinase was tested by molecular absorption spectrometry at 340 and 546nm in a Cobas c701 analyzer (Roche Diagnostics). Reference values are 0–4.5μKat/L.
The study included a dynamic FSH and LH stimulation test after the administration of 84.6mmol (100mg) of intravenous gonadorelin (GnRH). Few reference studies are available reporting normal gonadotropin levels in subjects with normal gonadal function. A classic study12 reported a two to ten-fold increase above normal LH levels after 100μg of GnRH, while FSH increase is less evident. Another study13 reported for males aged 18–65 years with normal gonadal function basal normal LH levels ranging from 0.4 to 6.9IU/L and FSH levels ranging from 0.4 to 5.8IU/L, while levels seen 60min after stimulation with 100μg of GnRH were 4.6–14.5IU/L and 2–5.2IU/L, respectively.
In all reported cases of suspected KD, DNA molecular analysis was performed using a polymerase chain reaction (PCR) of the polymorphic region of the AR gene. The number of CAG triplets present in the polymorphic region of exon 1 of the AR gene, located in the X chromosome, is measured. For this, DNA is extracted from total EDTA blood with Qiagen Mini Spin Blood columns. Subsequently, PCR amplification of the polymorphic region of the AR gene is performed from total genomic DNA. Amplification is performed with Taq polymerase (Ecotaq) using the program 95¿5′+35 X (95¿45″+68¿40″+72¿1′10″)+(72¿10′) in an Applied Biosystems GeneAmp 2700 thermal cycler.
This PCR is analyzed by electrophoresis in 2% agarose gel. The number of triplets in the sample is determined based on size. Values less than 35 triplets (up to 300bp) are considered normal, and those higher than 36 are considered pathological.
Case 1This was a 41-year-old male patient with no pathological history (PH) of interest, except keratoplasty. He had been diagnosed at 37 years of age based on neurological clinical signs consisting of proximal weakness in the lower limbs (LLs). Examination found proximal paresis, as well as areflexia, tongue atrophy, and progressive facial weakness. The patient currently maintains autonomy, with free handling function and walking capacity.
He had experienced gynecomastia and decreased body hair since puberty. The patient had experienced no voice or muscle changes, but reported decreased libido and sexual potency over the previous 3 years. He had no offspring, but no fertility study had been performed.
Genetic tests showed 60 CAG triplet repeats, which is in the range considered pathological (36–88 CAG repetitions).
In the study of first-degree relatives, the mother was diagnosed as carrying the mutation, while the maternal grandfather had experienced a similar neurological picture but had not been examined.
Physical examination: weight (W), 84kg; height (H), 1.69m; body mass index (BMI) 29.4kg/m2; blood pressure, 114/62mmHg. No goiter. The patient showed no eunuchoid proportions, but had decreased body hair and bilateral gynecomastia, which was more marked on the left side. Testicular volume: right testis (RT) 12cc and left testis (LT) 10cc.
Case 2This was a 58-year-old male patient with a PH of high blood pressure (HBP) treated with drugs, mild untreated dyslipidemia, and kidney stones. He had been diagnosed at 48 years of age based on neurological clinical signs and symptoms of cramp, asthenia, fatigue, and generalized weakness.
His pubertal development had been normal, with no gynecomastia. He reported androgenetic alopecia with neither changes in shaving frequency nor decreased body hair. He had noticed a gradual decrease in strength of voice and testicular volume in recent years, as well as decreased libido over the previous 15 years. The patient reported sexual impotence from the onset of the neurological symptoms.
The genetic study showed 50 CAG triplet repeats.
No genetic study had been made of his first-degree relatives, and he had no children despite never having used contraceptive methods during 35 years with a stable partner. He had never had a fertility evaluation.
Physical examination: W 76kg; H 1.75m; BMI 26.3kg/m2; BP 150/95mmHg. No goiter. No gynecomastia. Facial and body hair of male distribution. Testicular size: RT/LT 20/12cc.
Case 3This was a 66-year-old male patient with a PH of HBP treated with drugs. The neurological clinical signs and symptoms had started at 52 years, including LL weakness and fatigue with fasciculations and frequent falls. His symptoms had progressed making walking difficult, and necessitating the use of a wheelchair.
He had had bilateral gynecomastia since childhood, and no change had occurred after the onset of the neurological symptoms. The secondary sexual characteristics appeared late. He had consulted a doctor in adolescence, but no precise diagnosis was made, and his pubertal development was delayed. He reported a loss of voice strength and erectile dysfunction in parallel to the start of the neurological symptoms, but no decrease in libido.
The genetic study revealed 60 CAG triplet repeats.
No genetic study had been performed in parents or siblings. His two daughters were studied and found to carry the mutation. One of these had two sons in whom genetic testing ruled out the condition.
Physical examination: W 81kg; H 1.75m; BMI 26.4kg/m2; BP 140/50mmHg. No goiter. The patient had mild gynecomastia mainly in the left breast. Male distribution of facial and body hair. Testicular size: RT/LT 20/15–20cc.
Case 4A 70-year-old male patient with a PH of HBP and dyslipidemia on drug treatment and obstructive sleep apnea syndrome treated with continuous positive airway pressure. He had been diagnosed at 62 years of age for neurological symptoms of distal weakness in the upper limbs and paresthesia. A physical examination showed areflexia, atrophy of intrinsic hand muscles and distal apallesthesia in the LLs. The patient had had changes in strength of voice and mild dysphagia since diagnosis.
He reported normal pubertal development with no gynecomastia or other evidence of hypogonadism.
A prostate tumor was diagnosed at 64 years of age and treated with radiotherapy. Since then, the patient reported a decreased libido, which had not existed before this specific treatment.
The genetic study showed 63 CAG triplet repeats.
No genetic tests had been performed in first-degree relatives. The patient had five children (three male and two female) and one granddaughter, in all of whom the mutation had been ruled out.
Physical examination: W 73.6kg; H 1.64m; BMI 27.4kg/m2; BP 120/60. No goiter. Minimal right gynecomastia. Male distribution of facial and body hair. Examination of testes: RT/LT 20/20cc. Left inguinal hernia and inguinal scar from right inguinal hernia surgery.
DiscussionKD is a neurodegenerative disorder associated with the abnormal expansion of the CAG triplet in the gene encoding for AR.5,14
KD is a condition that occurs late. The mean age at symptom onset in our series was 49.3 years (37–62 years), similar to that reported in the literature, ranging from the second to the sixth decades of life.3,4,7,9,15,16 Cases starting in adolescence have been reported. These initially cause signs of hypogonadism such as gynecomastia, microorchidism, oligospermia, or azoospermia.9
Neurological involvement consists of the characteristic signs of involvement of the lower motor neuron, such as proximal or distal muscle weakness, muscle atrophy, facial, perioral and limb fasciculations, dysarthria, or dysphagia.17 Initial symptoms include tremor, cramps, and muscle weakness.17 Signs of upper motor neuron disease such as hypereflexia or spasticity have not been reported.3
The clinical neurological spectrum is very wide. Phenotypical data may range from an isolated laboratory change, such as elevated serum creatine kinase (CK) levels secondary to muscle atrophy, to more severe forms with bulbar muscle involvement which may require artificial ventilation and the placement of gastrostomy catheters for feeding.3,6
In our series, the main starting symptom leading patients to seek medical help was muscle weakness and fatigability of LLs, causing severe walking difficulties in one of the cases. In addition to LL weakness, the predominant neurological symptoms included cramps, tongue atrophy, and variable bulbar involvement, recognized as characteristic findings of the disease.3 One of the patients experienced painful distal hypesthesia concomitant with muscle weakness symptoms, which has been reported in less than half the cases in other studies.1,3,18
Endocrine findings in our patients consisted of gynecomastia, decreased body hair, testicular size reduction, decreased libido, and sexual impotence.3,9,19,20
Two of the patients had had bilateral gynecomastia since childhood and puberty, which represents a frequency similar to the 70% reported by Dejager et al.9 This is considered to be the most common endocrine manifestation.3,20 Some patients may develop these endocrine symptoms, and particularly gynecomastia, even earlier than the neurological clinical signs.9
Decreased libido and erectile dysfunction evolved in parallel to the neurological symptoms in two of the cases, while in the other two patients clinical symptoms appeared after endocrine changes. Decreased testicular size with no signs of sexual ambiguity such as hypospadias or micropenis was found in two cases.
Data from the hormone tests performed on our series of patients are summarized in Table 1. The hormonal test results of the first patient were normal. Two patients had increased levels of serum total testosterone (T) and SHBG, with decreased serum free testosterone fraction.9 According to the literature, serum T levels may be normal or elevated,9,16,21–23 with high levels even at an advanced age16; a positive correlation may be found between T and SHBG, which may be inappropriately increased.3,9,20 Serum estrogen (E2) levels were found to be increased in the last three cases. This increase may have been due to testosterone aromatization to estrogens.2,9,22
Gonadotropins were increased in the second case, while LH was slightly elevated in the fourth patient. A GnRH stimulation test performed in the last three patients showed LH hyperresponse. On the other hand, the second and third patients also had FSH hyperresponse, which was however not seen in the last patient. LH levels reported in the literature may be increased or not suppressed. LH response to GnRH stimulation may be exaggerated despite normal serum testosterone levels, reflecting partial androgen resistance.9
High serum LH levels result from the resistance of the hypothalamus and pituitary gland to the negative feedback mechanism by sex steroids due to androgen resistance. LH therefore stimulates increased testosterone and estradiol production by Leydig cells.11,24 Serum FSH levels were normal in three of our four patients, in agreement with previous reports.22,23
ASI, the product of LH and testosterone, has been suggested as a good marker of androgen resistance that reflects the impact of negative feedback on the hypothalamic-pituitary-testicular axis. It is thought that ASI may be a helpful marker for identifying patients at risk of mild forms of androgen resistance caused by AR mutations. Mean ASI in our series was 384.93IU×nmol/L2. In a prior study of 22 patients with confirmed KD, mean ASI was 166IU×nmol/L2 (range, 20.3–364IU×nmol/L2),9 higher than that found in a control group of healthy, fertile men (mean, 36IU×nmol/L2; range, 3.5–88IU×nmol/L2) or in other control groups taken from older studies (54IU×nmol/L2; range, 6.7–138.7IU×nmol/L2).23
Dejager et al.9 found a significant correlation between the size of CAG triplet expansion, ASI, and SHBG.9 This correlation was not seen in our series.
High CK levels have been reported in patients with clinical involvement. The first case in our series had CK levels eight times greater than normal.19
The endocrine picture of KD is a part of partial androgen insensitivity syndrome (PAIS), which includes the partial loss of secondary sexual characteristics such as gynecomastia, decreased facial hair and shaving frequency, testicular atrophy, decreased libido, erectile dysfunction, and oligospermia/azoospermia causing impaired fertility.11,25
AR is a nuclear hormone transcription factor responsible for androgen action in target tissues including seminal vesicles or prostate, among others, with a central role in male fetus virilization and spermatogenesis. AR is also expressed in motor neurons of the spinal and cranial nerves.5 AR is located on the long arm of the X chromosome (Xq11-12) and consists of eight 8 exons.3,16,26 The first exon, encoding the N-terminal region with a significant role in transactivation, encompasses the CAG nucleotide sequence, which in turn encodes the polyglutamine region in the corresponding AR protein.5,27,28 Variations in this polyglutamine chain have been associated with KD EK.2,8
The mechanisms underlying neurodegeneration in KD have not been fully elucidated. Expansion of the AR gene triplet repeat is likely to result in dysfunctional proteins with long polyglutamine sequences which, when translocated to the nucleus, produce nuclear polyglutamine inclusions and dysregulation of transcriptions.29 AR expression is particularly abundant in motor neurons, where androgens play a significant role in axonal regeneration.30
While normal individuals have 9–36 copies of the CAG triplet,3,20 affected males and female carriers have a pathological expansion of the region to 40–62 repeats3,16,26 which causes a gain in protein function and results in neurotoxicity.3,17,26 Experimental and clinical data support the hypothesis of a gain of neurotoxic functions as the molecular basis of the disease.31
Different studies have reported a positive correlation between expansion size and disease severity. A lower age at the onset of gynecomastia has also been correlated to greater androgen resistance.9 Several reviews agree that a negative correlation exists between expansion size and the age of onset of the disease.3,9,16,32,33 Other literature reviews have also established a direct relationship to the progression rate,23 but this however has not been supported by other studies.16 In our series, the patient with the highest number of copies was not diagnosed at an earlier age than the other cases, nor did his disease follow a worse course.
Mutations affecting the AR gene may be classified into two main categories: mutations that cause an expansion of the CAG triplet (a gain in neurotoxic function of the AR carrying the elongated polyglutamine region) and do not affect sexual differentiation but cause KD, and mutations leading to receptor function loss.28 The latter mutations cause sexual differentiation disorders in individuals with the 46 XY genotype, grouped together as androgen resistance syndrome (ARS).5 ARS is subdivided into two phenotypes based on the severity of androgen resistance: complete androgen resistance (complete androgen insensitivity syndrome or CAIS) and partial androgen resistance (partial androgen insensitivity syndrome or PAIS).11,34,35
Subjects with CAIS are characterized by having a female phenotype with female external genitalia, a short vagina, and an absence of structures derived from Wolffian ducts such as the uterus and Fallopian tubes. They also have adequate breast development in puberty with an absence of pubic or axillary hair.5,10,34–36
PAIS is an incomplete form of ARS with a wide clinical presentation spectrum ranging from external genitalia of predominantly female appearance to ambiguous genitalia or an essentially male phenotype with micropenis, perineal hypospadias, or cryptorchidism (a phenotype formerly included as Reifenstein syndrome).5,10,22,34,35
A third variant also reported in the literature, mild androgen insensitivity syndrome (MAIS), encompasses healthy males with poor virilization symptoms with several grades of gynecomastia, sparse pubic hair, and impotence. Spermatogenesis may or may not be impaired in this group. Thus, individuals affected may have different degrees of fertility.10,11,34 For some authors, KD could be included in this subtype.10
The four cases reported here may be considered as mild PAIS or MAIS; two of the patients even have children. However, it is a limitation of our study that genetic confirmation of ARS in the reported patients was not available.
AR is expressed in most prostate tumors,37 an association which suggests that AR may be a genetic predictor of individual susceptibility to prostate cancer.37 In these androgen-dependent tumors, progression, cell growth, and survival depend on AR regulation.33
Epidemiological clinical studies have established that a reduction in CAG triplet expansion to less than 22 copies directly correlates to a diagnosis of prostate cancer at an earlier age, with an increased risk and a greater predisposition to a more advanced tumor stage.33,38,39
One patient with KD in our series was subsequently diagnosed with prostate cancer, and a molecular study found a total of 63 copies of the CAG triplet. This result contrasts with the findings previously reported for isolated prostate tumors.
KD is diagnosed based on clinical history, physical examination, hormone tests, neurological diagnostic tools such as evoked potentials, electromyography, transcranial magnetic stimulation, and genetic studies.6 Diagnosis is confirmed by showing an expansion of CAG triplet repeats in the AR gene through genetic testing.3 CAG triplet expansion was confirmed in the patients in our series, all of whom had more than 36 copies.
The vast majority of patients have a positive family history, but approximately one-third of those diagnosed do not know of other probable relatives affected.17 Genetic tests allow for accurate diagnosis on an individual basis and for genetic counseling as part of the treatment.3,6
There is no established treatment for KD other than symptomatic management.3,5,6 Patients may receive vitamin E, vitamin B complex, or physical therapy as part of their treatment.3,6 Hormone replacement using testosterone or analogs could even reversibly worsen symptoms.3,5,40 Trials based on androgen deficiency have been conducted in mice using drugs such as the GnRH antagonist, leuprorelin, or the 5-alpha reductase inhibitor dutasteride.41 This prevents the nuclear translocation of aberrant AR,42,43 conferring protection against the toxic accumulation of AR mutant protein and suppressing any manifestations of neuromuscular impairment. However, randomized, placebo-controlled clinical trials have not shown significant effects on neurological functions such as muscle strength or swallowing.40,44
Only some patients required ventilatory support in advanced disease stages, but overall life expectancy is slightly compromised.3
ConclusionIn conclusion, we report four cases of KD confirmed by molecular study with a neurological and endocrine picture consistent with other series. A diagnosis of KD should be considered in any adult male with progressive muscle weakness and associated endocrine changes, with or without family history.
Conflicts of interestThe authors state that they have no conflicts of interest.
Please cite this article as: Valera Yepes R, Virgili Casas M, Povedano Panades M, Guerrero Gual M, Villabona Artero C. Enfermedad de Kennedy y resistencia parcial androgénica. Descripción de 4 casos y revisión de la literatura. Endocrinol Nutr. 2015;62:224–230.
