




La insulinorresistencia (IR) conlleva la insulinemia >1.000UI/dl para cifras normales de glucosa, y su prevalencia en la infancia oscila entre el 3,0 y el 8,4%1.
Su etiopatogenia puede ser primaria, por mutación génica o secundaria a obesidad y/o lipodistrofia2.
La IR hereditaria puede clasificarse en poligénica de severidad moderada, característica de la diabetes tipo 2 o monogénica, asociada a síndromes que presentan un fenotipo con obesidad centrípeta o lipodistrofia, o a receptoropatías hereditarias aisladas3, tales como:
Leprechaunismo o síndrome de DonohueDefecto autosómico recesivo completo de unión insulina-receptor, que produce una pérdida de la acción de la insulina. Se manifiesta con hipoglucemias precoces posprandiales, hiperinsulinemia, hiperlipidemia, crecimiento intrauterino retardado, dismorfias, disminución de tejido graso subcutáneo y masa muscular, macrogenitalismo y ovarios con quistes. Mortalidad frecuente antes de los 2 años de vida por infecciones intercurrentes4.
Síndrome de Rabson-MendenhallDefecto incompleto de unión insulina-receptor. Incluye excesiva producción de insulina y dificultad para eliminarla de la circulación. Es autosómico recesivo y el genotipo es predictor del fenotipo5. Presenta hiperinsulinemia, hiperglucemia posprandial seguida de hipoglucemia precoz, cetoacidosis constante, hiperlipidemia, retraso del crecimiento, distrofia muscular, dientes displásicos y prematuros, hipertrofia gingival e hiperplasia pineal. Su mortalidad está relacionada con su tendencia a la cetoacidosis y las complicaciones microvasculares4,6.
Síndrome de insulinorresistencia tipo ADisminución de la unión insulina-receptor y de su señalización. El fenotipo queda marcado por la resistencia a la insulina, mutaciones genéticas y los factores ambientales asociados5. Se manifiesta mediante intolerancia a la glucosa, hiperandrogenismo, talla alta y constitución corporal delgada y musculosa4.
La insulina actúa de forma diferente en los distintos tejidos. Los síntomas que pueden estar asociados a la IR según gravedad de la mutación son5–7:
- –
Acantosis por el efecto tóxico insulínico en la piel.
- –
Hiperandrogenismo por sobreproducción de testosterona premenstrual.
- –
Pubertad adelantada por la sinergia de gonadotropinas e insulina.
- –
Hipertrigliceridemia e hígado graso, por la unión selectiva al posreceptor hepático de insulina para producir síntesis y secreción grasa en el hígado.
- –
Seudoacromegalia, por sobrecrecimiento de tejidos laxos debido a una mutación posreceptor.
- –
Alteración del crecimiento lineal.
- –
Hipoglucemias posprandiales con hiperglucemias reactivas secundarias
- –
Obesidad centrípeta o lipodistrofia, por déficit de leptina o mutación de la diferenciación y regulación adipocitaria, fomentando la distrofia muscular.
La disminución de la actividad de la tirosina quinasa y la unión disminuida de insulina a los receptores son reversibles con la pérdida de peso6.
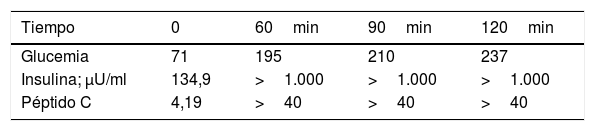
Presentamos el caso de un varón de 8 años con poliuria, polidipsia, hiperglucemia (319mg/dl) y glucosuria, sin cetonemia, de una semana de evolución. Se trata de un niño pretérmino que no presentó crecimiento intrauterino retardado. Entre los antecedentes familiares destaca madre con cáncer de mama, diabetes gestacional y posterior diabetes mellitus tipo 2, en tratamiento con antidiabéticos orales; y abuelo con diabetes mellitus tipo 2. A la exploración presenta peso 41,5kg (p53), talla 141cm (p56), IMC 20,3 (p52) y ausencia de acantosis nigricans. Analíticamente muestra anticuerpos anti-GAD, anticuerpos anti-insulina, anticuerpos anti-IA2 y estudio del gen GCK y HNF1A negativos (HNF4A no fue estudiado por no estar disponible en nuestro hospital). Péptido C basal: 4,19ng/ml, medido mediante quimioluminiscencia. Inicia tratamiento insulínico que suspende al año. Se pierde seguimiento, y a los 5 años tras el diagnóstico reaparece en consulta con HbaA1C del 7,7%, hiperglucemias posprandiales mantenidas, hipoglucemias preprandiales esporádicas y acantosis nigricans, a pesar de haber mantenido IMC normal desde el diagnóstico (IMC 21,23kg/m2). Presenta curva de glucemia-insulina-péptido C con insulinemia >1.000μU/ml realizada mediante radioinmunoensayo (tabla 1). Se estudiaron en el ADN de leucocitos de sangre periférica los 22 exones del gen INSR, con secuenciación directa (3.700 ABI), mostrando una alteración heterocigota en el exón 20 (p.Asp1177Glu, C.3531C>G), misma mutación que porta la madre y el abuelo (madre IMC 19,7kg/m2; abuelo IMC 22,9kg/m2). Es una mutación localizada en la cadena beta, compatible con un síndrome de insulinorresistencia tipo A, actualmente en tratamiento con metformina, con buen control glucémico (HbA1C 6,5%).
El síndrome de insulinorresistencia tipo A presenta una moderada-severa IR en ausencia de obesidad o lipodistrofia, al igual que nuestro paciente. Debido a su clínica sutil, sobre todo en la infancia, este síndrome se encuentra infradiagnosticado en la población pediátrica4.
El diagnóstico de IR se realiza mediante una buena anamnesis, controles glucémicos y analíticos, y estudio genético4.
La insulinorresistencia tipo A se relaciona con mutaciones del gen INSR, mostrando en nuestro caso una mutación no descrita, con buena correlación genotipo-fenotipo de acuerdo a los estudios in silico5,7. Se trata de una mutación heterocigota localizada en la cadena beta del INSR, que ha sido asociada en estudios funcionales con un fenotipo de IR más leve, como la que acontece en los familiares de primer y segundo grado de nuestro caso índice7.
Para disminuir los niveles de insulinemia se emplean sensibilizadores de insulina como tratamiento de primera elección, como en nuestro caso. En ocasiones es necesario el uso de insulina. Por otro lado, la IGF1 recombinante mejora la supervivencia en formas neonatales4.
Asimismo, cabe destacar en el caso presentado el antecedente de cáncer de mama materno, ya que la resistencia insulínica ha sido relacionada con el desarrollo de cáncer, debido a la similitud de los receptores de insulina con los receptores del factor de crecimiento8–10. El aumento de receptores IGF1 e IGF2 produce cambios en la dimerización del receptor (isorreceptores A y B), y una cascada de señales activadoras muy potentes que aumentan la proliferación de endometrio y mama mediante la activación de esteroides sexuales, a la vez que disminuyen la respuesta metabólica y la apoptosis.
Se ha comprobado una alteración del ratio de isorreceptores A:B en tumores con un aumento de isoforma A y una disminución de isoforma B, que se ha relacionado con el nivel de proliferación, pudiéndose usar como biomarcadores de pronóstico y respuesta al tratamiento, ya que los portadores de una mayoría de isorreceptor B son resistentes a tamoxifeno, con una mayor proliferación celular y metástasis, así como productores de mutagénesis y carcinogénesis por aumento de radicales libres y daño del ADN10, mientras que el predominio de subtipo A se corresponde con una buena respuesta al tratamiento y una menor proliferación9,10.
Finalmente, el aumento de tejido adiposo conlleva un aumento de células inflamatorias que activan dicho sistema inflamatorio y favorecen la producción de tumores8.
Como reflexión, sería importante más estudios que reforzaran estas teorías.
