






Los tumores neuroendocrinos hipofisarios (TNEH) constituyen, junto con otros tumores de la región selar, un 15-25% de las neoplasias intracraneales. En 2017, la Organización Mundial de la Salud propuso una nueva clasificación de los TNEH. La principal innovación respecto a la clasificación del 2004 fue la recomendación de incluir en la evaluación inmunohistoquímica de los TNEH la determinación de los factores de transcripción de las 3 líneas celulares hipofisarias: Pit-1, Tpit y SF-1. Adicionalmente, se han propuesto otras clasificaciones clínico-patológicas con capacidad predictora del comportamiento tumoral durante el seguimiento. Ante estos cambios, procede adaptar el conocimiento generado durante los últimos 15 años a la práctica diaria del tratamiento y seguimiento de los TNEH en los centros de excelencia de patología hipofisaria. El presente documento recoge el posicionamiento de la Sociedad Española de Endocrinología y Nutrición (SEEN) y la Sociedad Española de Anatomía Patológica (SEAP) sobre la clasificación de los TNEH y la información que el patólogo debe proporcionar al clínico para facilitar el tratamiento y seguimiento de estos tumores.
Pituitary neuroendocrine tumors (PitNETs) constitute, together with other tumors of the sellar region, 15-25% of intracranial neoplasms. In 2017, the World Health Organization proposed a new classification of PitNETs. The main innovation with respect to the 2004 classification was the recommendation to include in the immunohistochemical evaluation of PitNETs the determination of the transcription factors of the 3 pituitary cell lineages: Pit-1, Tpit and SF-1. Additionally, other clinicopathological classifications with a predictive capacity of tumor behavior during follow-up were proposed. Given these changes, it is appropriate to adapt the knowledge generated during the last 15 years to the daily practice of the treatment and monitoring of PitNETs at the Centers of Excellence in Pituitary Pathology. This document includes the positioning of the Spanish Society of Endocrinology and Nutrition (SEEN) and the Spanish Society of Pathology (SEAP) on the classification and denomination of the PitNETs and the information that the pathologist should provide to the clinician to facilitate the treatment and monitoring of these tumors.
Los tumores hipofisarios neuroendocrinos (TNEH)1, junto con otros tumores de la región selar, constituyen un 15-25% de las neoplasias intracraneales1-5. Su incidencia ha aumentado de forma progresiva y paralela al uso de técnicas de imagen cerebral y craneal cada vez más sensibles6,7. Aunque en el 85-90% de los casos se trata de tumores adenohipofisarios, existen otras lesiones de la región selar, como las neoplasias mesenquimales, meníngeas o de origen neural, las metástasis de múltiples orígenes y los quistes o procesos inflamatorios2-5.
Históricamente, los TNEH se han denominado adenomas debido a su habitual comportamiento indolente respecto a otras neoplasias y tumores neuroendocrinos de otras localizaciones. Recientemente, el International Pituitary Pathology Club, en su decimocuarta reunión celebrada en Annecy en noviembre de 2016, propuso sustituir el término adenoma por el de tumor hipofisario neuroendocrino (TNEH)1. Este término reflejaría mejor, según los autores del documento, las similitudes entre los tumores adenohipofisarios y los tumores neuroendocrinos de otros órganos y el comportamiento agresivo de algunos de los primeros. Sin embargo, esta propuesta ha sido motivo reciente de controversia8,9.
El debate terminológico se extiende también al uso generalizado del acrónimo genérico NFPA (del inglés non-functioning pituitary adenomas). Todos los subtipos de TNEH hormonalmente activos tienen homólogos silentes que se han agrupado bajo el acrónimo NFPA. El término adenoma hipofisario silente o TNEH silente describe los tumores adenohipofisarios que expresan una o más hormonas adenohipofisarias o sus factores de transcripción en la inmunohistoquímica (IHQ) pero no secretan hormonas en concentraciones clínicamente relevantes5,10,11. Sin embargo, ambos términos han sido utilizados de manera indistinta en la literatura para referirse al mismo tipo de tumor.
La reciente clasificación de la OMS de 2017 agrupa los TNEH en las 3 líneas celulares de la adenohipófisis de las que proceden mediante la determinación IHQ de los correspondientes factores de transcripción hipofisarios (FTH). Este agrupamiento facilita una aproximación diferente al diagnóstico patológico de los TNEH. En concreto, ha permitido reducir de forma drástica el porcentaje de tumores nulos y establecer una clasificación más precisa de los tumores plurihormonales10,12. Además, se ha propuesto que en el informe del patólogo figuren también datos clínicos (tumor funcionante/silente; presencia y grado de invasión radiológica) y datos de actividad proliferativa (MIB1-LI/Ki-67, número de mitosis y p53). El objetivo final ha sido diseñar una clasificación clínico-patológica de los TNEH con capacidad pronóstica de su comportamiento13-15. Esta propuesta ha sido respaldada por el European Pituitary Pathology Group16. Sin embargo, no todos los patólogos están de acuerdo, pues consideran que dicho informe solo estaría al alcance de centros de excelencia con disponibilidad de neuropatólogos especializados en enfermedad hipofisaria. Por el contrario, sostienen que es suficiente con que el informe del patólogo recoja datos macroscópicos, de tinción de hematoxilina-eosina, de inmunotinción de las hormonas adenohipofisarias y del MIB1-LI/Ki-6717.
En cualquiera de los casos, el informe del patólogo es esencial para la correcta planificación del tratamiento y seguimiento de los TNEH. Por tanto, los patólogos deben formar parte de los equipos clínicos multidisciplinares que atienden a pacientes con enfermedad tumoral hipofisaria.
Los recientes cambios introducidos por la clasificación de la OMS en 20172 y las controversias terminológicas con relación a los tumores neuroendocrinos y en el contenido del informe del patólogo justifican este documento. El objetivo es establecer el posicionamiento del Área de Conocimiento de Neuroendocrinología de la Sociedad Española de Endocrinología y Nutrición (SEEN) y de la Sociedad Española de Anatomía Patológica (SEAP) sobre la clasificación y denominación de los TNEH y la información que el patólogo debe proporcionar al clínico (endocrinólogo/neurocirujano) para facilitar el tratamiento y seguimiento de estos tumores.
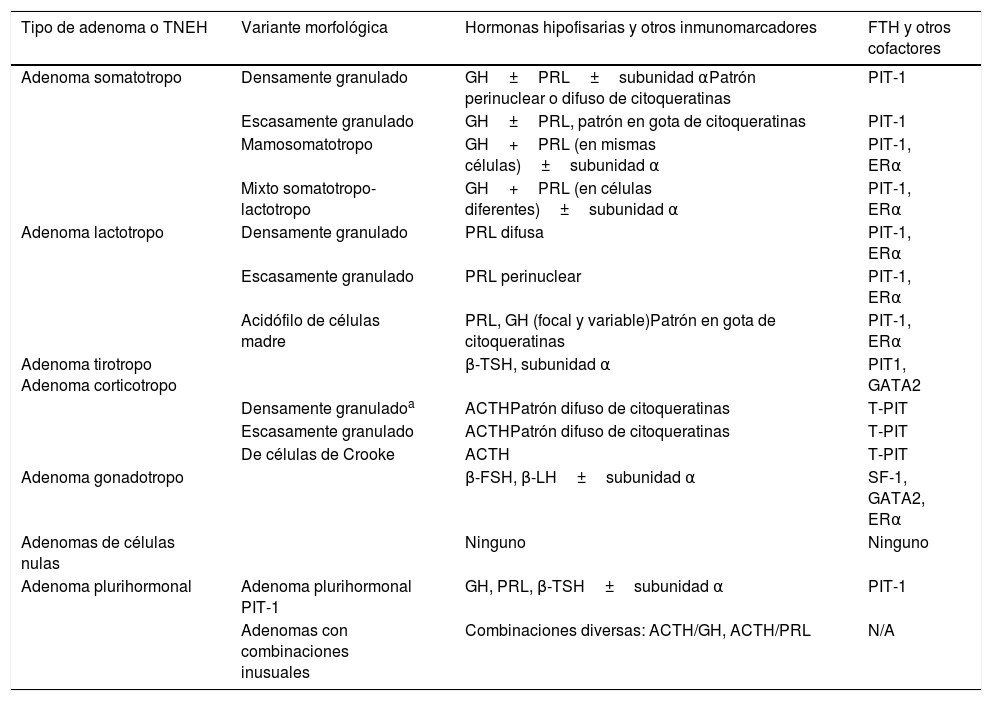
La nueva clasificación de los tumores hipofisarios neuroendocrinos de 2017: ¿cuáles son sus aportaciones?En 2017 se publicó la cuarta edición de la clasificación de tumores de los órganos endocrinos de la OMS2, con el objetivo de servir de referencia para todos los especialistas implicados en su diagnóstico y tratamiento. En esta edición se ha tenido en cuenta la expresión de los FTH de las líneas celulares hipofisarias, junto a las características estructurales y alteraciones genéticas de los TNEH (tabla 1). Como principales novedades se señalan las siguientes:
- a)
Se recomienda determinar los FTH de las 3 líneas celulares hipofisarias: el factor de transcripción 1 específico de la hipófisis (Pit-1), el factor de transcripción T-PIT y el factor esteroidogénico-1 (SF-1).
- b)
Se elimina el término de adenoma atípico, propuesto en la clasificación del 2004. En cambio, se identifican subtipos de alto riesgo, caracterizados por mayor capacidad de invadir, recidivar y metastatizar que los otros TNEH.
- c)
Se mantiene la recomendación de determinar el índice MIB1-LI/Ki-67 como marcador proliferativo y se relega la determinación de p53 a aquellos tumores con Ki-67 ≥ 3%.
- d)
Se incluye el blastoma hipofisario como nueva entidad.
Clasificación de los tumores neuroendocrinos hipofisarios
| Tipo de adenoma o TNEH | Variante morfológica | Hormonas hipofisarias y otros inmunomarcadores | FTH y otros cofactores |
|---|---|---|---|
| Adenoma somatotropo | Densamente granulado | GH±PRL±subunidad αPatrón perinuclear o difuso de citoqueratinas | PIT-1 |
| Escasamente granulado | GH±PRL, patrón en gota de citoqueratinas | PIT-1 | |
| Mamosomatotropo | GH+PRL (en mismas células)±subunidad α | PIT-1, ERα | |
| Mixto somatotropo-lactotropo | GH+PRL (en células diferentes)±subunidad α | PIT-1, ERα | |
| Adenoma lactotropo | Densamente granulado | PRL difusa | PIT-1, ERα |
| Escasamente granulado | PRL perinuclear | PIT-1, ERα | |
| Acidófilo de células madre | PRL, GH (focal y variable)Patrón en gota de citoqueratinas | PIT-1, ERα | |
| Adenoma tirotropo Adenoma corticotropo | β-TSH, subunidad α | PIT1, GATA2 | |
| Densamente granuladoa | ACTHPatrón difuso de citoqueratinas | T-PIT | |
| Escasamente granulado | ACTHPatrón difuso de citoqueratinas | T-PIT | |
| De células de Crooke | ACTH | T-PIT | |
| Adenoma gonadotropo | β-FSH, β-LH±subunidad α | SF-1, GATA2, ERα | |
| Adenomas de células nulas | Ninguno | Ninguno | |
| Adenoma plurihormonal | Adenoma plurihormonal PIT-1 | GH, PRL, β-TSH±subunidad α | PIT-1 |
| Adenomas con combinaciones inusuales | Combinaciones diversas: ACTH/GH, ACTH/PRL | N/A |
Modificado de la clasificación de la OMS de adenomas de la glándula hipofisaria2. FTH: Factores de transcripción hipofisarios.
La determinación IHQ adicional de las hormonas adenohipofisarias y de las citoqueratinas de bajo peso molecular permite identificar tipos de TNEH con variantes relacionadas con aspectos estructurales y predictivos de comportamiento.
La clasificación permite, además, identificar variantes homólogas silentes de los distintos subtipos funcionantes y reducir de forma significativa el porcentaje de tumores verdaderamente nulos.
Los tumores hipofisarios neuroendocrinos no funcionantesDesde la proliferación de técnicas de imagen muy sensibles, los TNEH no funcionantes son los tumores neuroendocrinos hipofisarios diagnosticados con mayor frecuencia, tanto en las series quirúrgicas como en las no quirúrgicas18. Los TNEH que no asocian un síndrome clínico hormonal reconocible se han agrupado, históricamente, bajo el acrónimo NFPA. Los NFPA son casi siempre macroadenomas y pueden presentar síntomas relacionados con el efecto masa (cefalea o alteraciones neuroftalmológicas), hipopituitarismo parcial o completo, hiperprolactinemia o ser descubiertos de manera incidental en una resonancia nuclear magnética cerebral. Cuando tras la cirugía se identifica la línea celular de la que deriva el tumor y este no asocia un síndrome endocrino sistémico, el tumor se denomina silente5,10,11. Por tanto, tras la cirugía, el término NFPA debería ser sustituido por el tipo celular del tumor que corresponda acompañado del adjetivo silente; por ejemplo, TNEH corticotropo silente, lactotropo silente, etc., de forma que se distinga de la variante funcionante respectiva. Alrededor de un 10% de los TNEH gonadotropos son funcionantes19. Por tanto, los adjetivos silente y funcionante también deberían aplicarse a este subtipo de TNEH. Los TNEH de células nulas, cada vez menos frecuentes conforme ha avanzado la precisión de las técnicas diagnósticas, deberían etiquetarse como tales. El espectro clínico de los TNEH muestra un continuo de secreción hormonal entre tumores funcionantes y completamente silentes20. La expresión completamente silente se ha referido a tumores hipofisarios de un determinado subtipo que cursan con concentraciones circulantes de la hormona correspondiente rigurosamente normales21. El término clínicamente silente se referiría a tumores que cursan con concentraciones hormonales circulantes algo elevadas de forma autónoma, pero sin expresar el correspondiente síndrome clínico21. Además, el estado funcional de un TNEH puede cambiar durante el seguimiento, con mayor frecuencia en el caso de los tumores de estirpe corticotropa22-24.
Sin duda, esta heterogeneidad terminológica ha condicionado los resultados y las conclusiones de muchas de las series publicadas. La tipificación patológica cada vez más precisa de los TNEH ha permitido identificar la identidad celular de más del 95% de los tumores hipofisarios y ha reducido el porcentaje de tumores nulos por debajo del 3%10,25,26. Por tanto, y debido al diferente comportamiento de los tumores procedentes de diferentes células hipofisarias, el término NFPA debería utilizarse para los TNEH que no asocian un síndrome endocrino exclusivamente antes de la cirugía y sustituirse después por el subtipo silente correspondiente.
El tumor agresivoLa definición de tumor agresivo es controvertida y también se ha utilizado de forma heterogénea en la literatura. En la clasificación de la OMS de 2004 se introdujo el concepto de adenoma atípico para referirse a aquellos adenomas que presentaban: 1) un índice mitótico >10 mitosis por campo; 2) un índice de proliferación Ki-67 ≥ 3% y 3) una fuerte tinción nuclear para p53. En la clasificación de la OMS de 2017 se elimina el término de adenoma atípico. En su lugar, se reconoce la existencia de subtipos de adenomas de elevado riesgo por su peor pronóstico durante el seguimiento: los lactotropos en varones, los corticotropos silentes y los adenomas de células de Crooke, los somatotropos poco granulados y los tumores plurihormonales Pit-1 silentes. Los plurihormonales Pit-1 sustituyen al antes denominado subtipo 3, un tumor caracterizado por su aparición en pacientes jóvenes y con mayor tasa de enfermedad persistente o recurrente durante el seguimiento27. Además, se continúa recomendando la inmunotinción de Ki-67 como criterio de proliferación y la de p53 solo cuando aquella es ≥3%.
Recientemente, la Sociedad Europea de Endocrinología28 ha definido al tumor hipofisario agresivo como «tumor grande, con invasión radiológica de las estructuras adyacentes y con un crecimiento inusual o con presencia de recurrencias pese a recibir un tratamiento óptimo». Sin embargo, con frecuencia, se confunden los conceptos de invasión y agresividad, ya que un tumor invasivo puede no ser agresivo mientras que todos los tumores agresivos son invasivos. Por tanto, ante la discrepancia de criterios clínicos y patológicos, es necesario diferenciar estos conceptos: 1) invasión: invasión radiológica o anatómica de los senos adyacentes a la silla turca, con independencia de la expresión de Ki-67; 2) proliferación: inmunotinción de MIB1-LI/Ki-67 ≥ 3%, con independencia de la presencia o no de invasión y 3) agresividad: tumor invasivo que recidiva o crece durante el seguimiento pese a recibir el correcto tratamiento, con independencia de los marcadores de proliferación.
¿Tumor de células nulas o tumor inmunonegativo?El tumor de células nulas es un tumor de células adenohipofisarias que no muestran una diferenciación celular específica en la IHQ de las hormonas adenohipofisarias o de los FTH. Por tanto, como sinónimos, se han propuesto los términos de adenoma inmunonegativopara hormonas hipofisarias o adenoma inmunonegativo para FTH29. Desde una perspectiva microscópica, está formado por células cromófobas o algo acidófilas, ordenadas con un patrón en forma de hoja. Se diferencian de los gonadotropinomas en que son menos frecuentes los patrones de crecimiento papilar o pseudopapilar. Su prevalencia ha cambiado mucho en las series recientes, a raíz del uso de anticuerpos más específicos frente a las hormonas hipofisarias15, del estudio IHQ de los FTH10 y de los estudios moleculares de expresión génica de las hormonas hipofisarias30,31 y de sus FTH26. Su prevalencia actual es inferior al 3% en todas las series mencionadas, cuando en un principio se habían descrito prevalencias entre un 5 y un 30% de las series quirúrgicas32,33.
Con las nuevas técnicas, la mayoría de los tumores considerados en principio como nulos se reclasifican como gonadotropinomas (la mayor parte expresan SF-1) o, con menor frecuencia, como corticotropinomas silentes34,35. Dado que los tumores verdaderamente nulos y los corticotropinomas silentes pueden comportarse de forma más agresiva que los gonadotropinomas36,37, tiene gran importancia su correcta identificación. Debido a la inmunonegatividad para las hormonas hipofisarias y para los FTH, el diagnóstico es de exclusión y la recomendación actual es denominarlos tumores inmunonegativos en lugar de tumores nulos.
Estudio anatomopatológico de los tumores hipofisarios neuroendocrinosEl estudio diagnóstico de enfermedad hipofisaria debe ser efectuado por un patólogo. Si es posible, según las características del centro, por un patólogo o neuropatólogo con contrastada experiencia en enfermedad hipofisaria. Se recomienda reservar parte del tejido tumoral para su criopreservación y almacenamiento en los biobancos, junto con muestras de sangre periférica para completar los estudios somáticos con estudios germinales. Se recomienda también la creación de colecciones de tumores neuroendocrinos en los respectivos biobancos que puedan interactuar en red.
Información clínica que debe conocer el patólogo antes del estudioEl diagnóstico anatomopatológico se realiza en un contexto clínico y, por ello, es indispensable que el patólogo reciba la información de contexto necesaria. En consonancia con el European Pituitary Pathology Group16, los autores recomiendan que el patólogo tenga acceso a la información recogida en la tabla 2.
Información que el clínico (endocrinólogo/neurocirujano) debe proporcionar al patólogo junto a la muestra de tejido para analizar
| Tipo de muestra | • Biopsia intraoperatoria• Tejido tumoral final• Hipófisis normal• Otros |
| Sospecha diagnóstica | • Síndrome clínico endocrino• No funcionante |
| Antecedentes familiares | • Neoplasia endocrina múltiple, adenoma pituitario familiar aislado, síndrome de Carney, feocromocitoma/paraganglioma |
| Tamaño tumoral | • Máximo diámetro en milímetros |
| Tipo de intervención realizada | • Transesfenoidal, microscópica, transcraneal |
| Características radiológicas | • Invasión de senos cavernosos, esfenoidal o de clivus• Intensidad en T2 |
| Grado de extirpación | • Completa, parcial, biopsia |
| Tratamiento previo | • Cirugía (reintervención)• Radioterapia (convencional, estereotáxica, radiocirugía)• Farmacológico: análogos de somatostatina 1.ª y 2.ª generación, agonistas dopaminérgicos, pegvisomant, ketoconazol |
Se recomienda que el material biológico sea remitido en fresco y sin fijador de forma inmediata tras su extracción para evitar la isquemia del tejido3,4,38. Idealmente se remitirá en una gasa humedecida en solución salina para evitar su disgregación en un recipiente lleno de solución. Debe remitirse al servicio de anatomía patológica todo el tejido obtenido y evitar al máximo la manipulación en el área quirúrgica. Antes de su fijación, el patólogo remitirá parte de la muestra al biobanco correspondiente para su criopreservación.
Análisis macroscópico y procesamiento del tejidoEn el análisis macroscópico se debe describir el número de fragmentos y su volumen aproximado. El patólogo se responsabiliza de la selección de tejido que se destine a técnicas especiales o al biobanco, siempre haciendo prevalecer las necesidades para el diagnóstico. Es recomendable un control microscópico de la muestra antes de congelarla, algo que puede hacerse de forma rápida con una muestra citológica por aplastamiento de un pequeño fragmento38.
Cuando procede hacer un diagnóstico intraoperatorio, para evitar la pérdida de tejido y la aparición de artefactos que se producen en las secciones por congelación, se recomienda una citología por extensión o aplastamiento de un pequeño fragmento3,4,39,40.
Estudio microscópicoEl tejido tumoral para el estudio diagnóstico se fijará en formalina neutra tamponada (entre 6 y 48 h). No se deben utilizar fijadores mercuriales o basados en ácido pícrico (Bouin)3,4,16,38,40. La evaluación inicial se hace con tinción de hematoxilina-eosina, para confirmar que se trata de un TNEH2-4,16,34,38,39. Se determinará el tipo tintorial, las características celulares (grado de pleomorfismo), arquitectura, grado de fibrosis, necrosis, signos de hemorragia o cualquier otro parámetro morfológico de interés. Se informará de la presencia de tejido hipofisario normal, si formase parte del material biológico remitido. Si existieran dudas de que el material biológico correspondiera a la glándula hipofisaria normal en lugar de al tumor, se hará una tinción para reticulina. Esta técnica es especialmente recomendable en el caso de la enfermedad de Cushing si no se ha identificado tumor en la MRI. Se cuantificará el número de mitosis en 10 campos (40×)2,16,39.
Estudio inmunohistoquímicoEl estudio inmunohistoquímico permitirá caracterizar el tumor y determinar variables de interés pronóstico de respuesta al tratamiento farmacológico2,16,34,41,42. Se debe tener en cuenta que no existen estudios comparativos respecto al rendimiento diagnóstico según los anticuerpos utilizados, sistema de visualización, cuantificación o puntos de corte que definan la positividad. Por tanto, cada laboratorio deberá definir y validar sus procedimientos. Aun así, los estudios de concordancia y validación entre la IHQ y el estudio molecular que se han publicado recientemente30,31,43 indican que las positividades en la IHQ inferiores al 5% deben ser interpretadas con cautela a la hora de tipificar un determinado subtipo tumoral. Afortunadamente, la determinación por IHQ o técnicas moleculares de los FTH permitirá una adecuada tipificación de estos casos dudosos.
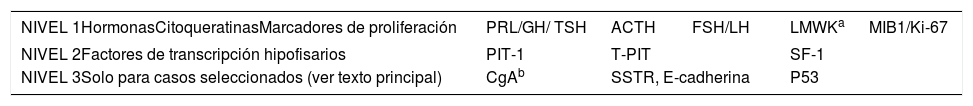
Inmunotinción de las hormonas hipofisarias y de los factores de transcripción hipofisarios de las líneas celulares hipofisariasRecientemente, el European Pituitary Pathology Group16 ha propuesto una estratificación por pasos de los estudios inmunohistoquímicos (tabla 3).
Recomendaciones multinivel del estudio inmunohistoquímico de los tumores neuroendocrinos hipofisarios de acuerdo con las recomendaciones del European Pituitary Pathology Group16
| NIVEL 1HormonasCitoqueratinasMarcadores de proliferación | PRL/GH/ TSH | ACTH | FSH/LH | LMWKa | MIB1/Ki-67 |
|---|---|---|---|---|---|
| NIVEL 2Factores de transcripción hipofisarios | PIT-1 | T-PIT | SF-1 | ||
| NIVEL 3Solo para casos seleccionados (ver texto principal) | CgAb | SSTR, E-cadherina | P53 | ||
ACTH: hormona corticotropa; FSH: hormona foliculoestimulante; GH: hormona del crecimiento; LH: hormona luteinizante; LMWK: citoqueratinas de bajo peso molecular; MIB1/Ki-67: índice de proliferación de MIB1; PIT1: factor de transcripción 1 específico de la hipófisis; PRL: prolactina; SF-1: el factor esteroidogénico-1; SSTR: receptores de somatostatina; TPIT: el factor de transcripción t-box; TSH: hormona estimulante de la tiroides.
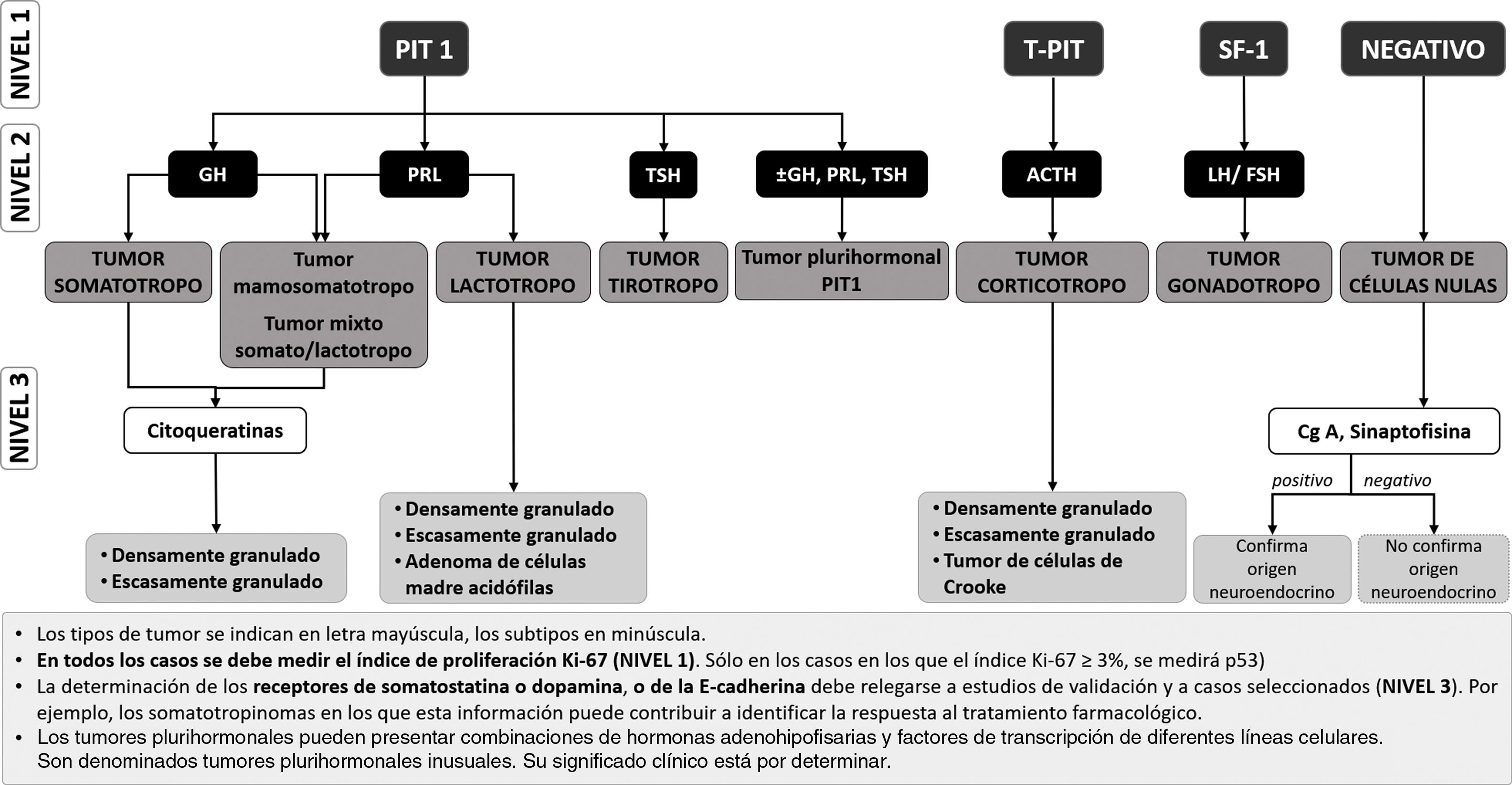
Sin embargo, en aquellos centros donde se puedan medir los FTH de forma rutinaria, se propone una secuencia diagnóstica diferente, que se recoge en la figura 1. Este esquema sería más eficiente en la identificación precisa de la mayor parte de los TNEH y supondría un importante ahorro de anticuerpos y de tiempo. Los tumores plurihormonales no se identificarán con tanta precisión con esta estrategia, pero suponen menos del 1% de todos los TNEH25.

Propuesta de algoritmo diagnóstico de los subtipos de TNEH sobre la base de la determinación en un primer escalón de los FTH hipofisarios.
PIT-1: factor de transcripción 1 específico de la hipófisis; T-PIT: el factor de transcripción t-box; SF-1: el factor esteroidogénico-1. ACTH: hormona corticotropa; GH: hormona del crecimiento; FSH: hormona foliculoestimulante; LH: hormona luteinizante; PRL: prolactina; TSH: hormona estimulante de la tiroides. Cg-A: cromogranina A.
La medición de la expresión de citoqueratinas de bajo peso molecular, en particular las 8 y 18, habitualmente identificadas con el anticuerpo CAM5.2 (CK8 y CK7), es útil para identificar de forma consistente algunas variantes de los subtipos de TNEH, en particular, los 2subtipos principales de somatotropinomas. Un patrón perinuclear difuso de citoqueratinas es diagnóstico de somatotropinoma densamente granulado, mientras que un patrón en gota con un porcentaje de cuerpos fibrosos >70% identifica al somatotropinoma escasamente granulado. Distinguir entre ambos e incluirlos en el informe del patólogo es relevante para el clínico, ya que los somatotropinomas densamente granulados presentan una mejor respuesta a los análogos de la somatostatina de primera generación44. Un patrón de expresión citoplasmática abundante se asocia también al TNEH corticotropo. Sin embargo, la mayoría de los adenomas negativos para citoqueratinas corresponden a gonadotropos16,34,39 y, por tanto, no estaría indicada su medición en este subtipo.
Evaluación de la proliferación: índice MIB1-LI/Ki-67Es importante estimar la proliferación celular mediante la cuantificación inmunohistoquímica del índice de tinción MIB1-LI/Ki-67, ya que este se ha considerado un posible factor pronóstico en los TNEH2,16,42 (tabla 3). La cuantificación con Ki-67 se realizará en 2de las áreas de la preparación con mayor número de núcleos teñidos (hot spots), considerando entre 500 y 1.000 células16, y se expresará en porcentaje sobre el número total de células tumorales. A diferencia de otros tumores neuroendocrinos, en los TNEH no se ha establecido un punto de corte que prediga de forma muy precisa el comportamiento agresivo del tumor2,16. Se acepta que un Ki-67 ≥3% está presente en la mayoría de los tumores agresivos28 y si excede el 10% debe considerarse la posibilidad de que se trate de una metástasis selar o de un carcinoma hipofisario45,46.
Otros marcadores pronósticos: número de mitosis e intensidad de inmunotinción de p53La cuantificación del número de mitosis en los TNEH no tiene la misma importancia que tiene en la gradación de los tumores neuroendocrinos del tracto gastrointestinal. El resultado se expresa por milímetro cuadrado, que equivale a 5 campos de gran aumento (40×) y es relevante la presencia de 2mitosis por cada 10 campos de gran aumento16,42. La positividad de p53 en el tejido tumoral es indicativa de mutaciones o inactivación del gen supresor TP53 y se considera marcador de comportamiento agresivo en muchos tumores sólidos47. Sin embargo, su importancia pronóstica en los TNEH es limitada2, por lo que su determinación solo se recomienda en aquellos tumores con un índice Ki-67 ≥ 3%, lo que ahorra costes y facilita una estrategia diagnóstica homogénea aplicable a todos los hospitales del país.
Otras determinaciones inmunohistoquímicasEn el caso de tumores nulos, puede ser de utilidad la inmunotinción de cromogranina A (CgA) y de sinaptofisina para certificar el origen neuroendocrino de la lesión y diferenciarla de otros tumores del área selar48.
Aunque diversas publicaciones señalan que la expresión del Pituitary Tumor Transforming Gene ayudaría a predecir la agresividad de los TNEH, esto no se ha confirmado en series grandes13. Por tanto, se desaconseja su determinación. El estudio de la expresión de receptores de la somatostatina puede ayudar a predecir la respuesta de los somatotropinomas y los NFPA al tratamiento con análogos de somatostatina de primera y segunda generación49-51. Además, recientes estudios han demostrado que la determinación de E-cadherina permite identificar con mayor precisión que la expresión de SSTR la respuesta a los análogos de somatostatina42,43,52. Sin embargo, los algoritmos de tratamiento de la acromegalia aún no contemplan la determinación IHQ sistemática de SSTR o de E-cadherina en la práctica clínica diaria. Por lo tanto, su determinación queda relegada, por el momento, a la investigación o a casos puntuales debidamente justificados2,16.
La determinación de metilguanina-metiltransferasa (MGMT), la enzima reparadora del ADN y que atenúa el efecto de la temozolamida en el tratamiento de los carcinomas y TNEH agresivos, se ha propuesto como un marcador de respuesta negativa a este fármaco. Datos recientes no han confirmado que la expresión de MGMT, su IHQ, la metilación del promotor de MGMT, los valores de índice proliferativo con Ki-67 o p53 sean predictores de la repuesta a temozolamida53. Además, no hay un consenso establecido con relación a cómo evaluar MGMT. En consecuencia, la determinación rutinaria de MGMT no está hoy justificada como predictor de respuesta.
Como alternativas al estudio de MGMT se ha propuesto la evaluación de proteínas reparadoras del ADN, en particular de MSH6, cuya expresión se ha asociado a respuesta a temozolamida54. La pérdida de inmunoexpresión nuclear de MSH6 y también de MSH2 se ha asociado a pérdida de eficacia al tratamiento con temozolamida. Sin embargo, los datos sobre su utilidad son muy limitados en la actualidad.
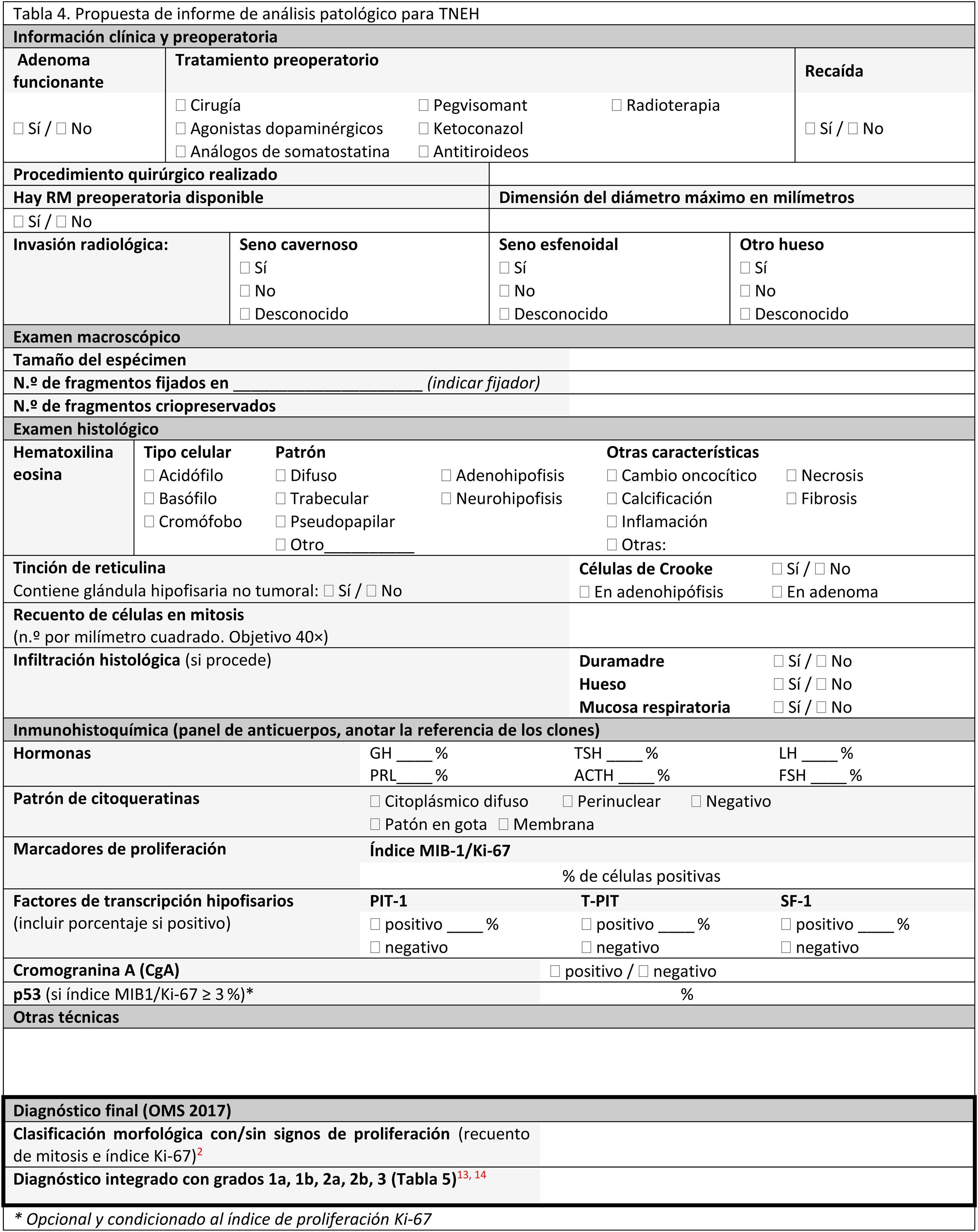
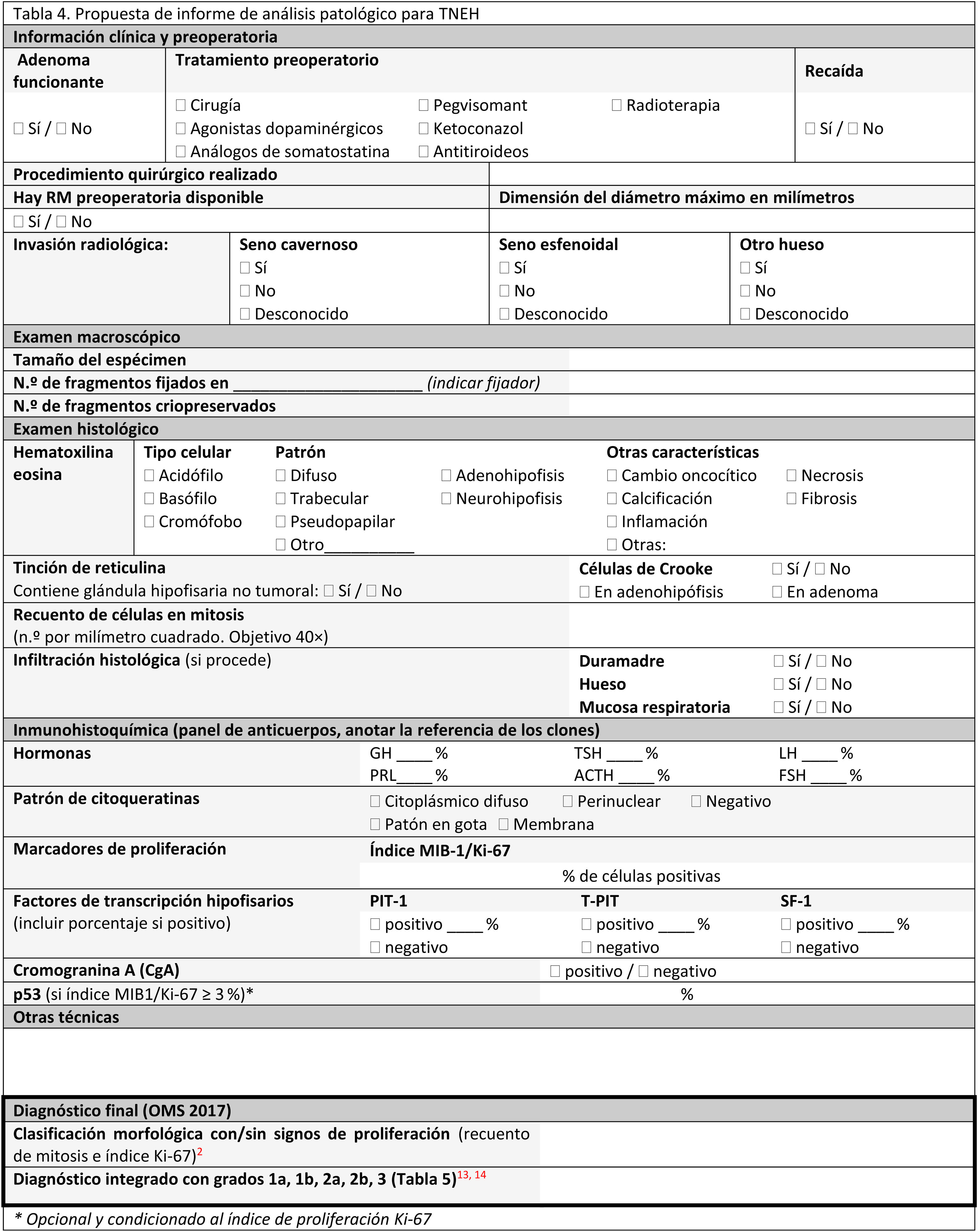
El informe anatomopatológicoAl igual que en otras áreas de la patología, se recomienda la utilización de modelos de informes anatomopatológicos estandarizados16,2,4,13,16,34,38,39,55,56. El informe debe contener: datos clínicos, datos radiológicos, datos de la intervención quirúrgica y datos de patología: microscopia e IHQ de las hormonas adenohipofisarias y, si está disponible, de los FTH (tablas 4 y 5).
Clasificación clínico-patológica de los tumores neuroendocrinos hipofisarios según Trouillas et al., 20139,14
| La clasificación se basa en 3 características: | |
| 1. Diámetro del tumor en micro (<10 mm), macro (≥10 mm) o gigante (≥40 mm) según estudio de RMN | |
| 2. Tipo de tumor según estudio inmunohistoquímico: GH, PRL, ACTH, FSH/LH y TSH | |
| 3. Grado tumoral basado en los siguientes criterios: | |
| Invasión: definida como signos histológicos o radiológicos (RMN) de invasión del seno cavernoso o esfenoidal | |
| Proliferación: considerada según al menos 2 de los 3 criterios: | |
| • Ki-67: >1% (fijador Bouin-Hollande) o ≥3% (fijador formalina) | |
| • Mitosis:n >2/10 campos de gran aumento | |
| • P53: positivo (>10 núcleos fuertemente positivos/10 campos de alta potencia) | |
| Grados de comportamiento tumoral establecidos según las características previas: | |
| Grado 1a | Tumor no invasivo |
| Grado 1b | Tumor no invasivo y proliferativo |
| Grado 2a | Tumor invasivo |
| Grado 2b | Tumor invasivo y proliferativo |
| Grado 3 | Tumor metastásico (metástasis cerebroespinales o sistémicas) |
Campo de gran aumento: 0,30mm2, aumento de 400×.
ACTH: hormona corticotropa; FSH: hormona foliculoestimulante; GH: hormona del crecimiento; LH: hormona luteinizante; PRL: prolactina; RMN: resonancia magnética; TSH: hormona estimulante de la tiroides.
En la descripción del estudio IHQ se han mencionado las limitaciones actuales de determinados marcadores, como el PTTG, para predecir agresividad o de los receptores de la somatostatina (clásicos SSTR1-5, y truncado sst5TMD4) o E-cadherina para predecir la respuesta de los somatotropinomas y NFPA al tratamiento con análogos de somatostatina. Todavía no hay evidencia consistente que los incluya en los algoritmos de tratamiento de la acromegalia para la elección del primer escalón de tratamiento médico. Así pues, por el momento, su uso queda restringido a investigación o a casos puntuales2,16.
Entre otros marcadores interesantes, los niveles de proteína RKIP se correlacionan de manera significativa con la respuesta de los niveles séricos de GH e IGF-1 y, como consecuencia, los bajos niveles de expresión de RKIP y la falta de inhibición de RAF-cinasa se asocian con una peor respuesta a análogos de la somatostatina en somatotropinomas57. La expresión del gen AIP es relevante en el diagnóstico de adenomas pituitarios familiares aislados y también en la respuesta terapéutica de los adenomas somatotrópicos58.
En los últimos años se han identificado otros marcadores de proliferación celular, apoptosis, angiogénicos, oncogenes, genes supresores de tumores, mediadores del ciclo celular, microARN y ARN largos no codificantes, que participan en la tumorogénesis, migración, proliferación y capacidad invasiva de los adenomas hipofisarios59.
Sin embargo, todavía no hay marcadores morfológicos confiables que predigan la recurrencia de un TNEH. Investigaciones recientes han permitido obtener nueva información sobre la génesis y el comportamiento biológico de estos tumores. Entre las moléculas exploradas se incluye la familia TACSTD (EpCAM, TROP2), neuropilina (NRP-1), senescencia inducida por el oncogén OIS, fascinas (FSCN1), genes asociados a la invasión (CLDN7, CNTNAP2, ITGA6, JAM3, PTPRC y CTNNA1) EZH2 y genes ENC1 y endocan60. Las técnicas moleculares, la bioinformática y las nuevas opciones farmacológicas son herramientas útiles para ampliar la comprensión de la naturaleza compleja de la tumorigénesis pituitaria51,59,61. Conforme se vaya constatando su utilidad clínica deberán ser incluidos en el informe patológico.
ConclusionesEl conocimiento de los TNEH se ha incrementado de forma exponencial durante los últimos años gracias a la mayor especificidad de los anticuerpos frente a las hormonas adenohipofisarias, la progresiva incorporación de la determinación de los FTH y los nuevos avances en biología molecular. En conjunto, estos avances permiten tipificar en la actualidad de forma correcta los diferentes subtipos de TNEH en más del 95% de los casos, lo que favorece su tratamiento y seguimiento. Además, la posibilidad de medir los factores de transcripción permite una estrategia de estudio secuencial patológico diferente a lo realizado hasta ahora. Si la estrategia propuesta en el presente artículo demuestra su eficacia, significará un importante ahorro en tiempo y coste de reactivos. Es necesario, pues, actualizar el informe que deben emitir los servicios de anatomía patológica para ayudar al clínico en el tratamiento óptimo de los TNEH.
FinanciaciónEl artículo fue financiado por la Fundación de la Sociedad Española de Endocrinología y Nutrición gracias un grant de Pfizer. Este documento ha recibido apoyo económico de Pfizer mediante una beca sin restricciones. Pfizer no ha participado en la redacción ni en su contenido.
Conflicto de interesesAP ha participado en ensayos clínicos relacionados con la enfermedad hipofisaria promovidos por Pfizer, Novartis e Ibsen, y en la difusión de conocimientos en simposios y reuniones patrocinados por Pfizer, Novartis e Ibsen. GM ha recibido honorarios como conferenciante de Novo Nordisk y Lilly y soporte para asistencia a congresos de Pfizer y Menarini. MPD ha recibido becas de apoyo a la investigación de Pfizer y Novartis, ha percibido honorarios como conferenciante de Ipsen y como asesor para Recordati. IAL, MAJ, OT y RML no tienen conflictos de intereses que declarar.
Los autores agradecen la colaboración de la Dra. Blanca Piedrafita, de Medical Statistics Consulting S.L., en la preparación y edición del manuscrito. Todos los autores contribuyeron en la redacción, realizaron revisión crítica de su contenido y aprobaron la versión final.
