




El síndrome de deficiencia de GLUT1 (GLUT1-SD), transportador de glucosa del sistema nervioso central, es una enfermedad metabólica congénita debido a las mutaciones en el gen SLC2A1; es considerada como una enfermedad rara o poco frecuente, por su baja incidencia en la población general de 1:90.000, habiéndose señalado más de 500 casos en la literatura1,2. El GLUT1-SD presenta trastornos del movimiento con distonía paroxística inducida por ejercicio, convulsiones y retraso mental. El diagnóstico se basa en la presencia de hipoglucorraquia, en ausencia de hipoglucemia, confirmándose mediante estudio molecular del gen SLC2A1. La dieta cetogénica es el tratamiento de elección para el GLUT1-SD3–7, proporcionando cuerpos cetónicos como única fuente de energía para la neurona. Presentamos el primer caso de GLUT1-SD y COVID-19 de la literatura. Mujer de 57 años de edad con diagnóstico tardío de GLUT1-SD, en seguimiento en los Servicios de Neurología y Endocrinología y Nutrición del Hospital Universitario Ramón y Cajal de Madrid, desde 2016. La clínica que presentaba al diagnóstico era: deterioro cognitivo, precisando para vivir un centro institucionalizado y tutorizada por una hermana, epilepsia y distonía progresiva paroxística, necesitando tratamiento con carbamazepina y levodopa/carbidopa. El estudio genético confirmó el diagnóstico de GLUT1-SD, al detectar la mutación p.Arg126Cys (p.R126C) en posición 376 (c.376C > T) del exón 4 del gen SLC2A1. La paciente es seguida en la Unidad de Enfermedades Metabólicas Congénitas del servicio de Endocrinología y Nutrición, llevando tratamiento con dieta cetogénica 2.5:1, con aporte de 1.500 kcal totales, con alimentos de bajo contenido en azúcares, aceite vegetal y MCT y suplemento de 100 mL cada 12 horas de Ketocal 4:1 LQ Multi Fibre. Este es un preparado líquido nutricionalmente completo, rico en lípidos, de bajo contenido en hidratos de carbono, con proteínas de suero suplementadas con aminoácidos y micronutrientes. La densidad energética del brick de 200 mL es de 300 kcal (1,5 kcal/mL), y contribuye a cubrir las necesidades calóricas proteicas.
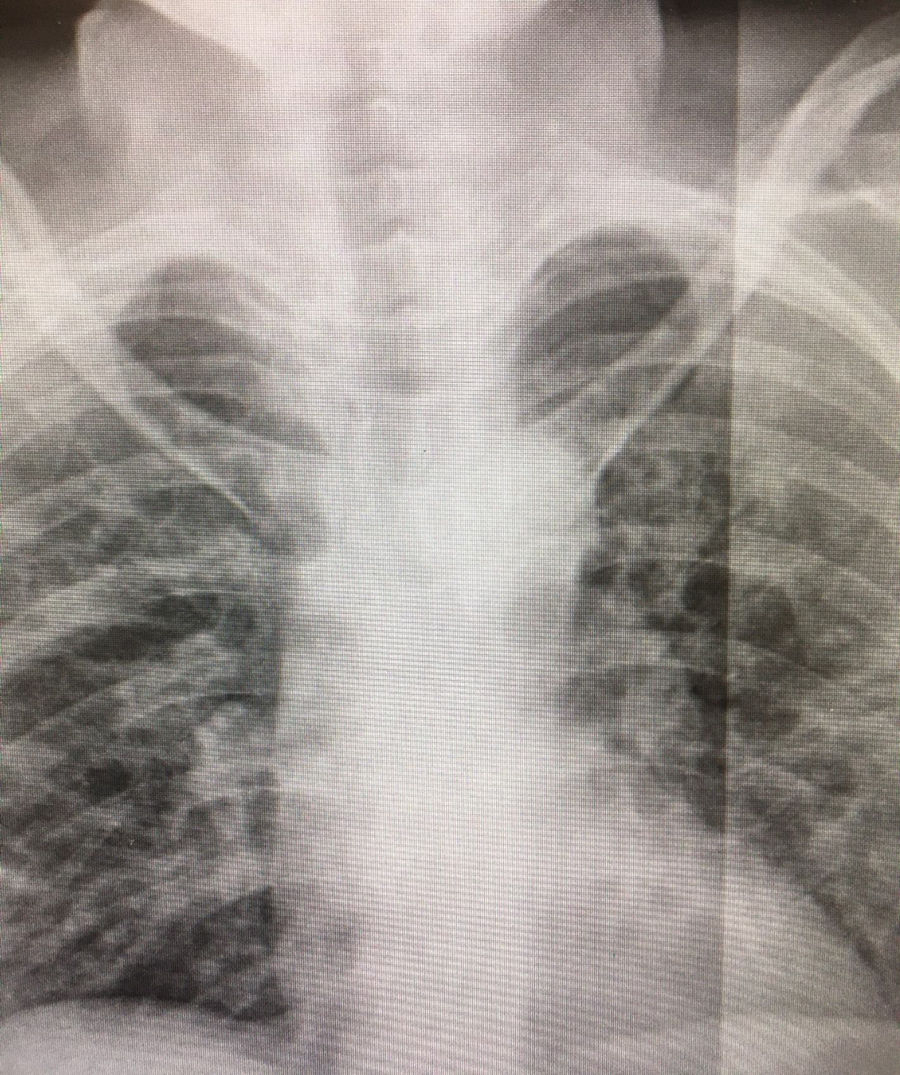
Con esta dieta presentó una notable mejoría del trastorno del movimiento, así como ausencia de nuevas crisis epilépticas, controlándose la dieta y el soporte nutricional mensualmente mediante correo electrónico y contacto telefónico con la residencia y tutora.La paciente acudió al Servicio de Urgencia del hospital por fiebre de 48 horas y disnea, presentando radiológicamente neumonía bilateral, típica de la COVID-19 (figura 1). La analítica al ingreso fue: glucosa 104 mg/dL, creatinina 0,62 mg/dL, TFG estimada (MDRD-4 IDMS) 99,93 mL/min, creatinquinasa 104 U/L, albúmina 3,08 g/dL, proteínas totales 6,2 g/d, AST/GOT 57 U/L (4-50), ALT/GPT 30 U/L, GGT 26 U/L, LDH 507 U/L (140-240), fosfatasa alcalina 53 U/L, proteína C-reactiva 129,6 mg/L (0-5), procalcitonina 0,11 ng/mL, troponina I 0,0 ng/mL, pH en gases 7,48, pCO2 31,0 mmHg, pO2 69,0 mmHg, HCO3 23,1 mM/L, % sO2c 94,8%, sO2m 97,4%, sodio en plasma 132,0 mM/L (135-145), lactato en gases 0,90 mM/L, calcio iónico 4,30 mg/dL, HB en Co-Ox. 10,3 g/dL (12-16), dímero D 540,00 ng/mL (0-500), hematíes 4,24 106/μL, hemoglobina 13,1 g/dL, hematocrito 35,9%, VCM 84,8 fL, HCM 30,9 pg, CHCM 36,4 g/dL, ADE 12,0%, plaquetas 312,0 103/μL, VPM 7,12 fL (7.5-11), leucocitos 7,14 103/μL, neutrófilos 4,67 103/μL, neutrófilos 65,40%, linfocitos 1,98 103/μL, tiempo de protrombina 12,30 sg, actividad protrombina 80,40%, INR 1,11, tiempo de cefalina 28,50 sg, fibrinógeno derivado 740,00 mg/dL (150-400). Ingreso por neumonía grave (CURB-65 2 puntos), siendo el test PCR de COVID-19 positivo. Se instauró tratamiento con Dolquine y Kaletra, ajustando la carbamazepina y manteniendo fluidoterapia con fisiológico al 0,9% y 500 mL de glucosa a 5% en 24 horas, así como dieta cetogénica 2.5:1 que tenía. Las distonías empeoraron en 24 h, por lo que se cambió a dieta cetogénica 4:1 exclusiva con Ketocal 4:1 LQ Multi Fibre cuatro bricks/día, 1.200 kcal. La paciente evolucionó favorablemente desde las 36 horas de esta dieta y fue dada de alta al domicilio con su hermana, con mejoría clínica, bioquímica, radiológica y negativización del test de PCR de COVID-19 a las tres semanas del ingreso. Desde entonces, la paciente sigue soporte nutricional ambulatorio por nuestra parte con dieta cetogénica 2.5:1 y evoluciona favorablemente con normalización de parámetros nutriciones glucosa 74 mg/dL, proteínas totales 7,5 g/dl, albúmina 4,67g/dL, prealbúmina 22 mg/dL, proteína ligada al retinol 4,3 mg/dL y proteína C reactiva 0,5 mg/dL.La paciente presentó la clínica, radiología y analítica con LDH, proteína C reactiva y dímero D elevados, como marcadores de la enfermedad y confirmación con test positivo PCR de COVID-19, y empeoró de su distonía, con la trasgresión de dieta al aumentar 25 g/día (500 cc glucosado 5%) el aporte de carbohidratos, y por tanto disminuir la cetogenicidad de la misma. Se ha referido que el estrés infeccioso puede descompensar a pacientes con GLUT1-SD5–8; en nuestro caso, la paciente presentó un discreto aumento de la ataxia sin crisis tónicas al iniciarse fluidoterapia, aunque limitada en glucosa desde su ingreso. Con una dieta cetogénica 4:1 completa exclusiva con cuatro bricks/día de Ketocal 4:1 LQ Multi Fibre consiguió normalizar la ataxia en 36 horas. El ser centro de referencia de enfermedades metabólicas congénitas, permitió a la familia ponerse en contacto con el Servicio de Endocrinología y Nutrición y poder iniciar el tratamiento específico al disponer de medios para el tratamiento de pacientes con errores congénitos del metabolismo9. Por tanto, debemos señalar dado lo excepcional del caso COVID-19 en paciente adulto con GLUT1-SD, este no supuso un factor de riesgo de complicación de COVID-19. Por último, destacar la importancia no sólo de la dieta personalizada, sino también el de disponer de suplementos específicos para la enfermedad metabólica en las unidades de nutrición que traten a pacientes con enfermedades metabólicas congénitas.

