





Multiple acyl-CoA dehydrogenase deficiency (MADD), also known as glutaric aciduria type II, is an autosomal recessively inherited metabolic disorder affecting the oxidation of fatty acids as well as the catabolism of branched-chain amino acids, lysine and tryptophan. MADD is caused by deficiency of either an electron-transfer flavoprotein (ETF, encoded by ETFA and ETFB) or an electron-transfer flavoprotein dehydrogenase (ETFDH, encoded by ETFDH).1
Patients with MADD have been classified into three groups: (1) neonatal onset without anomalies, (2) neonatal onset with anomalies and (3) mild or late-onset, with a wide range of clinical expression between the groups. Patients in the first group are often premature and present dysmorphic features with most of them dying in the first days of life. Patients in the second group do not present congenital anomalies but usually develop severe cardiomyopathy and die during the first weeks of life. The course and presentation in the third group, the late-onset patients typically include episodes of metabolic acidosis, non-ketotic hypoglycaemia and muscle weakness.2,3
The diagnosis of MADD consists in increased organic acid and acylglycine derivatives in the urine and medium- and long-chain acylcarnitines in the blood. Genetic analysis confirms the diagnosis.4
The treatment usually includes a high-carbohydrate, low-fat, low-protein diet associated with riboflavin and carnitine supplementation, without periods of fasting.4,5
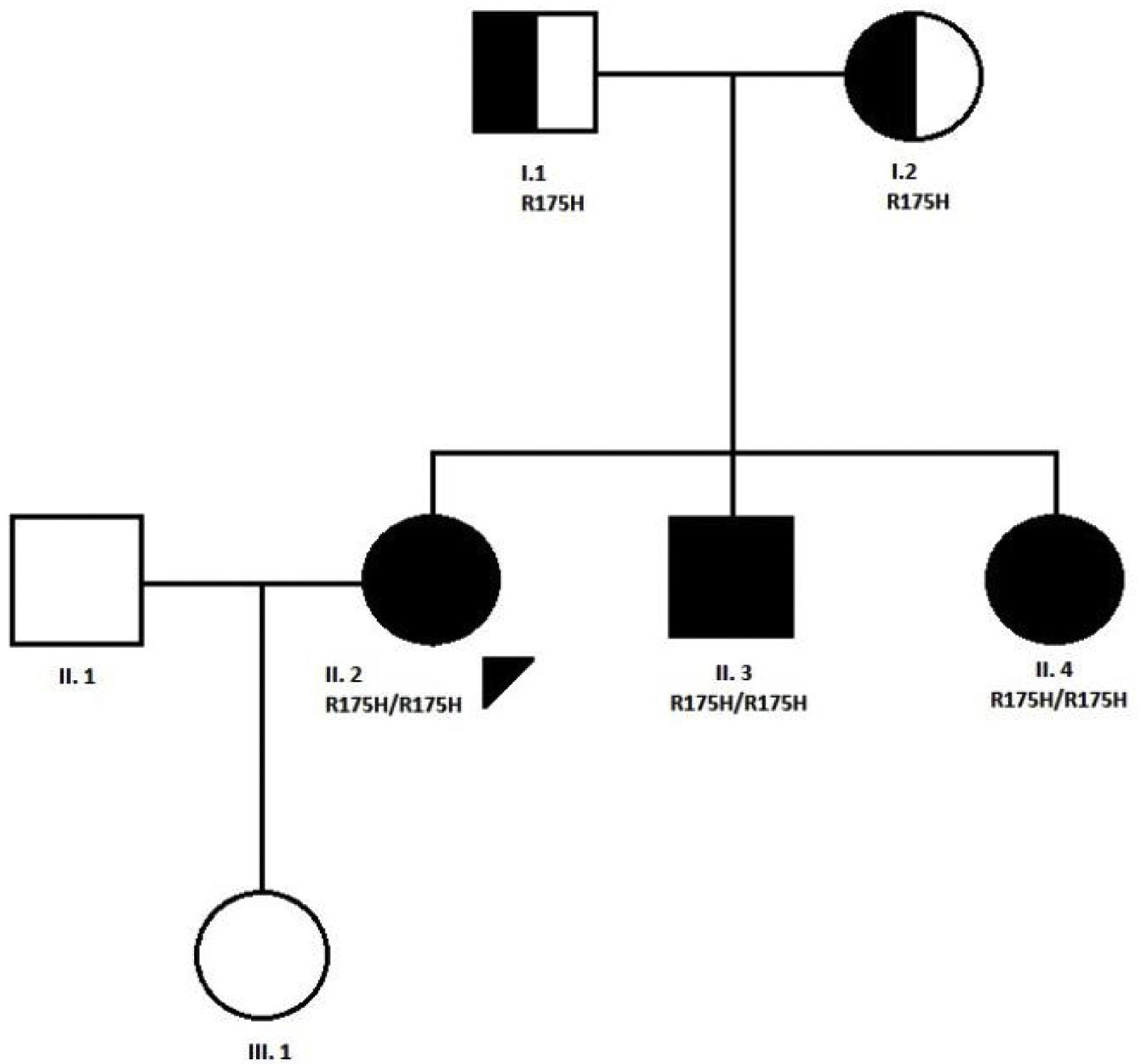
CaseWe present the case of a 31-year-old woman, diagnosed with MADD (mild or late-onset subtype) at the age of 10 years in the context of a family study because one of her brothers was hospitalised in the ICU in relation to a coma caused by metabolic acidosis and hypoglycaemia. Both of her parents were carriers of a mutation (R175H) in the ETFDH gene, and the three siblings were affected by the same condition. The patient is a product of non-consanguineous Spanish parents from the province of Ciudad Real. The paternal grandparents were first cousins (Fig. 1).

Pedigree of the family with MADD disease. The proband case is indicated with an arrow; circle and square symbols represent women and men, respectively; shaded symbols indicate the affected members; half-shaded indicate the carriers, Line 1 below symbols corresponds to the individual identification, line 2 indicates the mutation in the ETFDH gene, if present.
At the time of diagnosis she presented with high levels of acylcarnitines in her blood. Treatment with a diet restricted in fat and protein and associating supplementation of riboflavin and carnitine was started. Since the diagnosis to the present time the patient has been in good general condition, only suffering from occasional events of vomiting and muscle weakness (never requiring hospitalisations for metabolic decompensations).
Several echocardiograms have been performed throughout her life because of an increased risk of cardiomyopathy, which have all been normal.
At the age of 30, she became pregnant. She was not blood-related to her partner.
A normocaloric diet with most of the calories coming from carbohydrates (around 50/60%), similar to the diet she was already doing, was prescribed. The diet was divided into three main meals and periodic snacks, avoiding fasting periods of more than 4h to prevent hypoglycaemia. The treatment also included riboflavin (300mg/day) and l-carnitine (4g/day).
Usual obstetric controls in the first, second, and third trimester were performed, with an estimated foetal weight of 2.4kg (16% percentile) in the third trimester.
At the 39th week an elective caesarean was conducted to reduce the intrapartum risks (risk of metabolic decompensation and podalic presentation). During the caesarean and the postoperative period, she received intravenous carnitine (2000mg/day) and isotonic solution with 10% dextrose to prevent hypoglycaemia.
No complications during the procedure occurred and the patient remained clinically stable. Levels of glucose, creatine kinase, transaminases, lactic acid and ammonia were periodically measured and were within range limits.
The new-born was a phenotypically normal 2.5kg girl and had no problems during the first days of life. Breast feeding was initiated without incidents.
As the only complication she presented dacryocystitis when she was at 40 days, which required hospital admission and treatment with antibiotic therapy. From this moment to date no other incidents have occurred.
DiscussionOur patient, just like her brothers, had the mild or late-onset form of MADD, and as pregnancy is a stressful event which may produce metabolic decompensation (especially nausea and vomiting during the first trimester and labour itself) a strict follow-up in pregnant women with MADD is essential.
To our knowledge there are only two cases in the literature of a successful pregnancy in a woman with this condition. One is a 24-year-old woman with an atypical late-onset disease, because she had congenital anomalies (joint malformations), although it is possible that these were unrelated. This case presented several episodes of vomiting which required hospital admission and treatment with intravenous carnitine, dextrose and riboflavin during pregnancy. As with our patient an elective caesarean was performed, with no incident during the delivery and a very similar preventive treatment as in our case.6 The other case is a 19-year-old woman with late-onset MADD, with no decompensations during the pregnancy and delivery though caesarean without complications with the preventive treatment.7
In the absence of clinical guidelines for the care of pregnant woman with MADD this case reports entail important and helpful examples to guide the management of these patients.
