


To elucidate the pathogenetic mechanisms of a mutant P86S glucagon receptor (GCGR) in causing a novel human disease (Mahvash disease).
Material and methodEnhanced green fluorescent protein (EGFP)-tagged WT and P86S GCGR were expressed in HEK 293 or H1299 cells either transiently or stably. Receptor localization and internalization, and cell apoptosis were studied by fluorescence microscopy, and calcium signaling by Rhod-3 labeling. Gene expression was assayed by RT-PCR or Western blot. Cell fate was determined by live cell imaging.
ResultsUnlike WT GCGR, P86S was partially localized to the plasma membrane and partially in the cytoplasm as previously reported and did not undergo internalization upon glucagon treatment. P86S did not elicit calcium response after treatment with 1μM glucagon. Cells transiently expressing P86S exhibited more apoptosis than those expressing WT GCGR (18.3% vs 2.1%, P<0.05) but the X-box binding protein 1 mRNA cleavage, a marker of endoplasmic reticulum (ER) stress, was not evident, suggesting that the apoptosis did not result from ER stress. Cells stably expressing P86S did not exhibit apoptosis and a quarter of them harbored a novel inclusion body-like circular structure that was marked by P86S and ER residential proteins. These circular ER bodies were not seen in cells expressing WT GCGR or transiently expressing P86S and were not affected by treatment with proteasome inhibitor or microtubule depolymerizer, suggesting that they do not represent aggresome structures. The circular ER bodies could fuse and split to form new bodies.
ConclusionThe naturally-occurring P86S mutant GCGR exhibits abnormal receptor internalization and calcium mobilization, and causes apoptosis. The novel dynamic circular ER bodies may be adaptive in nature to nullify the toxic effects on P86S. These findings provide further insights into the pathogenetic mechanisms of Mahvash disease.
Conocer los mecanismos patogenéticos de un receptor del glucagón (GCGR) con la mutación P86S en una nueva enfermedad humana (enfermedad de Mahvash).
Material y métodoSe expresaron tipos silvestre y P86S del GCGR marcados con proteína verde fluorescente mejorada (EGFP) en células de HEK 293 o H1299 de forma estable o transitoria. Se estudió la localización e internalización del receptor, y la apoptosis celular mediante microscopio de fluorescencia, y la señalización del calcio mediante marcadores Rhod-3. La expresión génica se analizó mediante RT-PCR o Western blot. El destino de la célula se determinó mediante imágenes de las células vivas.
ResultadosA diferencia del tipo silvestre, la mutación P86S se localizó parte en la membrana plasmática y parte en el citoplasma como se ha notificado con anterioridad, y no se produjo internalización tras tratamiento con glucagón. La P86S no indujo respuesta de calcio después del tratamiento con 1μM de glucagón. Las células que expresaban transitoriamente P86S exhibieron más la apoptosis que aquella con GCGR silvestre (18,3% frente a 2,1%, p<0,05), pero no resultó patente la existencia de ARNm escindido en proteína de unión X-box 1, un marcador de estrés en el retículo endoplasmático (RE), lo que indica que la apoptosis no fue el resultado de estrés en el RE. Las células que expresan establemente la P86S no mostraron apoptosis y una cuarta parte de ellas albergaba la inclusión de una estructura circular corpusculoide nueva marcada por P86S y proteínas residentes del RE. Estos cuerpos circulares no se observaron en las células que expresaban GCGR silvestre o transitoriamente P86S y no se vieron afectados por el tratamiento con el inhibidor del proteasoma o despolimerizador microtubular, lo que sugiere que no representan estructuras agresivas. Los cuerpos circulares en RE podían fusionarse y dividirse para formar nuevos cuerpos.
ConclusiónLa mutación natural P86S de GCGR muestra una internalización anormal del receptor y de la movilización de calcio, y causa apoptosis. Los nuevos cuerpos circulares y dinámicos en el RE puede que sean una respuesta adaptativa para anular los efectos tóxicos sobre P86S. Estos hallazgos proporcionan nuevas perspectivas sobre la patogénesis de la enfermedad de Mahvash.
Naturally-occurring mutations of G protein-coupled receptors are uncommon but cause many human diseases.1,2 Inactivating mutations of G protein-coupled receptors result in loss of function of the normal signaling pathway mediated by the receptors. The mechanisms for loss of function by the inactivating mutations can be categorized into 4 classes of defective receptor function: receptor biosynthesis, trafficking to the cell surface, ligand binding, and receptor activation.2 The glucagon receptor (GCGR) belongs to the secretin receptor family of G protein-coupled receptors.3–5 GCGR is mainly expressed in liver and kidney. GCGR couples to Gs and Gq and increases cAMP and intracellular calcium levels, and undergoes glucagon-induced internalization.4–9 We have recently identified a novel human disease of hyperglucagonemia without glucagonoma syndrome, hypoglycemia, pancreatic α cell hyperplasia, and pancreatic neuroendocrine tumors associated with a homozygous inactivating mutation (P86S) of the GCGR (Mahvash disease).10,11 The mechanisms for pancreatic α cell hyperplasia and neuroendocrine tumors are not clear but appear to be loss of negative feedback inhibition on α cells by glucagon signaling. The P86S mutation is in the N terminal extracellular domain of GCGR. The homologous proline residue in the receptors of gastric inhibitory polypeptide (GIP) and glucagon-like peptide 1 (GLP-1), both highly homologous to GCGR, is critical for the binding with their respective ligands.12,13 The P86S mutant localizes partially to the plasma membrane and partially in the cytoplasm, binds glucagon with much lower affinity, and produces lower cAMP levels at physiological concentrations of glucagon but was able to achieve maximal cAMP levels at 100nM of glucagon. As the patient's phenotype is almost identical to that of mice lacking GCGR,14,15 it is likely that the P86S mutation causes the patient's unique condition.
To further characterize the P86S GCGR, we studied its internalization, calcium mobilization, effects on cell survival, and detailed subcellular localization by expressing enhanced green fluorescent protein (EGFP)-tagged WT or P86S GCGR in HEK 293 or H1299 cells, and we demonstrated that the P86S mutant has multiple abnormalities in receptor function. Moreover, the P86S mutant induces formation of a novel subcellular structure, dynamic circular endoplasmic reticulum bodies, which may be an important mechanism for cellular processing of the mutant GCGR.
Materials and methodsCell Culture, plasmids, transfection, and reagentsHEK 293 (from human embryonic kidney) and H1299 (from human non-small cell lung carcinoma) cells were grown in Dulbecco's modified Eagle's medium with 10% fetal bovine serum in a 37°C, humidified incubator with 5% CO2. HEK 293 cells were used in most experiments as they are derived from the kidney which normally expresses GCGR. H1299 cells were only used in calcium measurements because they did not apparently express GCGR intrinsically through preliminary experiments and HEK 293 cells did not load the calcium dye Rhod-3 AM well. Plasmids encoding enhanced green fluorescent protein (EGFP)-tagged human WT and P86S glucagon receptor (GCGR) and the parental plasmid pEGFP-N3 were described before.11 Cells were transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, EE.UU.). Stable HEK 293 cell lines expressing EGFP, WT GCGR-EGFP, or P86S-EGFP were established by incubating cells with 0.6mg/ml G418 after transfection. Thapsigargin was from Sigma (St Louis, IL, EE.UU.), LLnL from Calbiochem (La Jolla, CA, EE.UU.), and colcemid from Invitrogen.
Calcium analysisH1299 cells were incubated with 10μM Rhod-3 AM, PowerLoad, and 2.5mM probenecid (Molecular Probes, Eugene, OR, EE.UU.) in Hank's buffered salt solution (HBSS) for 45minutes, followed by incubation in HBSS with probenecid for another 45minutes. Loaded cells were placed in a confocal microscope incubator chamber and images were acquired with a TCS SP confocal microscope (Leica Microsystems, Heidelberg, Germany) using excitation/emission 550/580nm. Scanning rate was set to 7 frames per second for 200seconds.
Apoptosis assayHEK 293 cells were grown on coverslips for 2-5 days after transfection. Cells were fixed with 4% paraformaldehyde, permeabilized with 0.6% Tween 20, stained with Hoechst 33258 (1:10,000; Molecular Probes), and observed with a 40×objective. Green cells with apoptotic nuclear characteristics (i.e. nuclear condensation and fragmentation) were scored as apoptotic, which was confirmed by terminal dUTP nick-end labeling staining (TUNEL) (Roche Molecular Biochemicals, Indianapolis, IN, EE.UU.).16
RT PCR and Western blotTotal RNA was extracted with RNeasy kit (Qiagen, Valencia, CA, EE.UU.) and treated with DNase. RNA (2μg) was reverse-transcribed using SuperScript II Reverse Transcriptase (Invitrogen). Human XBP1 (X-box binding protein 1) was amplified by PCR using 100 ng cDNA and primer set GAAGCCAAGGGGAATGAAGT (forward) and ACTGGGTCCAAGTTGTCCAG (reverse) for XBP-117 and primer set for actin B (Real Time Primers, Elkins Park, PA). Western blot of EGFP-tagged WT or P86S GCGR was described previously.11
Immunofluorescent staining and fluorescence microscopyThese were performed as described.18 Briefly, HEK 293 cells expressing WT GCGR-EGFP or P86S-EGFP were fixed with 4% paraformaldehyde, and permeabilized with blocking buffer. Primary antibody, rabbit polyclonal anti-calregulin or anti-glucosidase II, goat polyclonal anti-ubiquitin (Santa Cruz Biotechnology, Santa Cruz, CA, EE.UU.), or mouse monoclonal anti-alpha tubulin (Calbiochem) was added at 1:50 in the blocking buffer and incubated for 2h. Cells were washed and incubated with appropriate rhodamine-labeled secondary antibody for 30min. EGFP fluorescence was directly visualized without staining. Immunostained cells were visualized on a TE200 inverted epifluorescence microscope (Nikon, Melville, NY, EE.UU.) equipped with relevant fluorescence filters. For live cell observation, HEK 293 cells stably expressing WT GCGR-EGFP or P86S-EGFP were grown in gridded flasks and the same groups of cells were directly visualized over time with green fluorescence filters.18
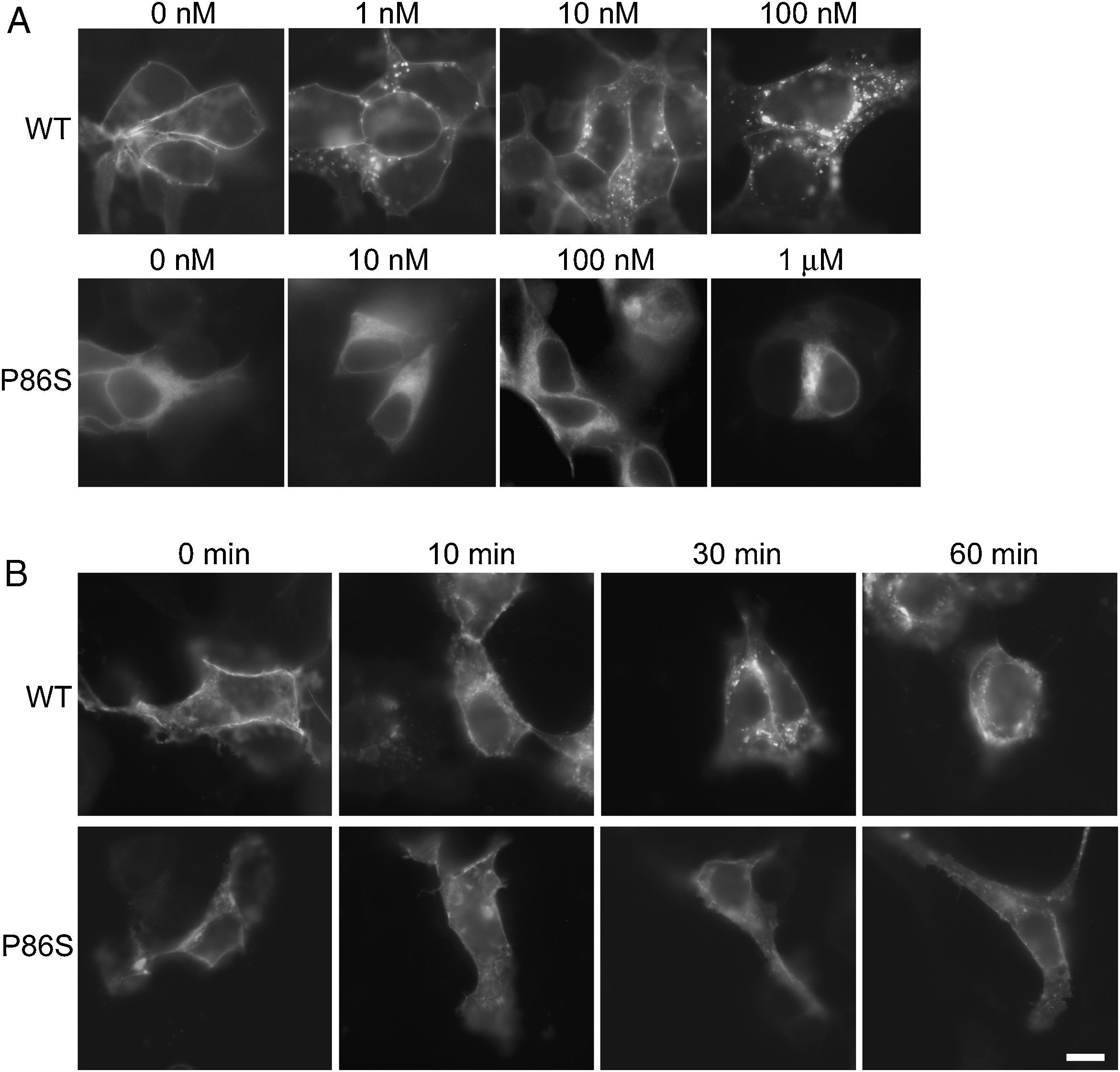
ResultsReceptor internalizationIn HEK 293 cells, WT GCGR was predominantly localized on the plasma membrane and underwent glucagon-induced internalization into intracellular vesicles in a glucagon dose-dependent manner (fig. 1A), in agreement with the results from Sachdev et al.9 Internalization of WT GCGR appeared to be complete after 30minutes of maximal glucagon stimulation (fig. 1B). P86S was partially localized to the plasma membrane and partially in the cytoplasma as previously reported.11 Cells with more cytoplasmic P86S localization were shown in Figure 1A while those with more plasma membrane localization were shown in Figure 1B. Regardless of the basal receptor localization, P86S failed to accumulate in intracellular vesicles in response to glucagon stimulation, even at 1μM (which stimulates maximal cAMP production by P86S11) for 1 hour. Thus, the P86S is defective in receptor internalization.
 GCGR internalization by increasing concentrations of glucagon. Two days after transfection with plasmids encoding WT GCGR-EGFP or P86S-EGFP, HEK 293 cells were treated with glucagon from 1nM to 1μM at 37C for 30minutes. Cells were fixed and representative cells shown. B) Time course of glucagon-induced GCGR internalization. HEK 293 cells were treated with 1μM glucagon for 10-60minutes. Note that untreated WT GCGR was always localized to plasma membrane while P86S was localized to cytoplasm in some cells (shown in A) or to plasma membrane in others (shown in B). P86S did not change localization upon glucagon stimulation. Bar, 10μm. EGFP: enhanced green fluorescent protein; GCGR: glucagon receptor.")
Internalization of WT GCGR and P86S. A) GCGR internalization by increasing concentrations of glucagon. Two days after transfection with plasmids encoding WT GCGR-EGFP or P86S-EGFP, HEK 293 cells were treated with glucagon from 1nM to 1μM at 37C for 30minutes. Cells were fixed and representative cells shown. B) Time course of glucagon-induced GCGR internalization. HEK 293 cells were treated with 1μM glucagon for 10-60minutes. Note that untreated WT GCGR was always localized to plasma membrane while P86S was localized to cytoplasm in some cells (shown in A) or to plasma membrane in others (shown in B). P86S did not change localization upon glucagon stimulation. Bar, 10μm.
EGFP: enhanced green fluorescent protein; GCGR: glucagon receptor.
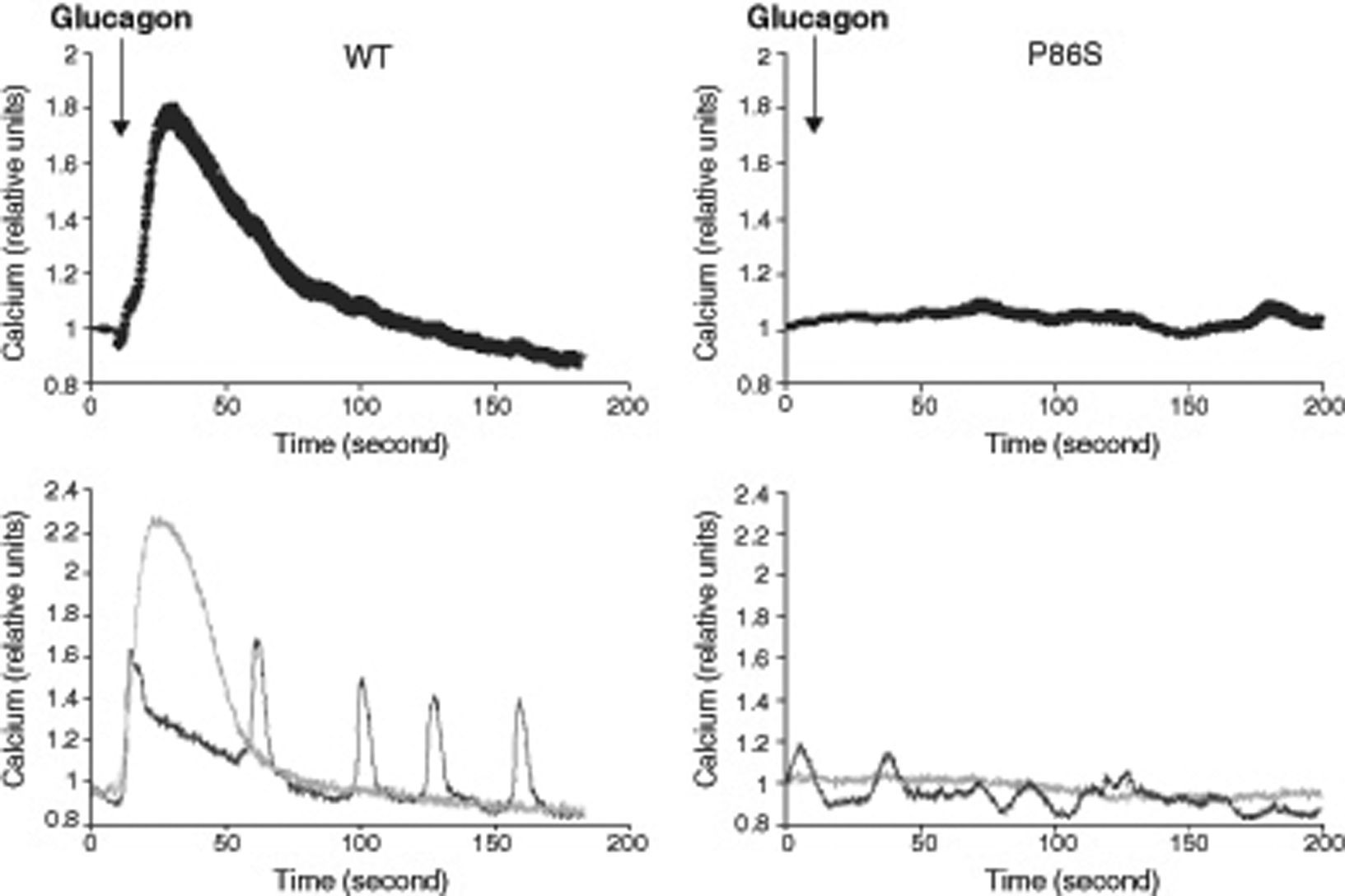
We used red calcium sensor Rhod-3 to study glucagon-stimulated calcium response of EGFP-tagged WT or P86S GCGR. In cells expressing WT GCGR, 1μM glucagon elicited a clear increase in intracellular calcium levels (fig. 2). Most cells exhibited a monophasic calcium response that largely tapered in 100seconds after glucagon stimulation. In a few cells, calcium levels began to oscillate after glucagon. In cells expressing P86S, no calcium increase was observed in response to glucagon stimulation. A few cells exhibited spontaneous low-amplitude oscillation of calcium levels which was not changed by glucagon.

Calcium signaling by WT GCGR and P86S. H1299 cells were transfected with plasmids encoding WT GCGR-EGFP or P86S-EGFP, loaded with Rhod-3 AM and stimulated with 1μM glucagon. Shown in upper panels were average Rhod-3 fluorescence levels±standard error of the mean before and after glucagon stimulation. Shown in lower panels were traces of 2 representative individual cells. P86S did not respond to glucagon with an increase in intracellular calcium.
EGFP: enhanced green fluorescent protein; GCGR: glucagon receptor.
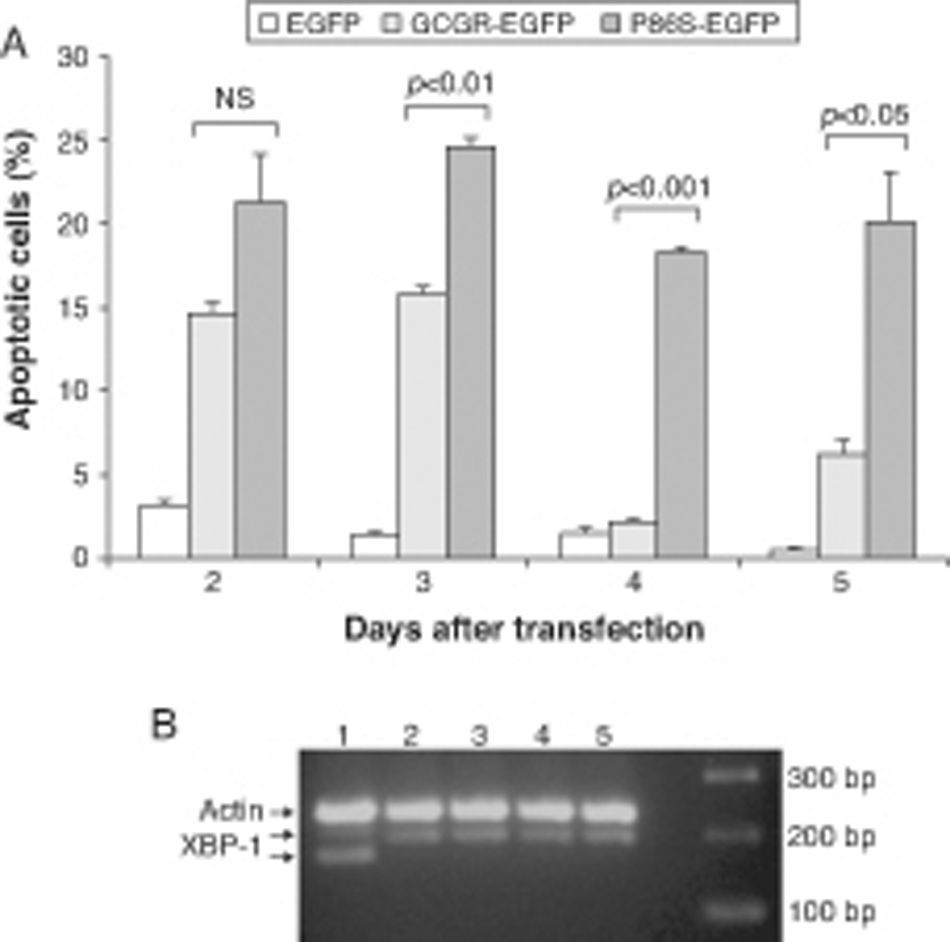
We measured apoptotic rate of HEK 293 cells expressing EGFP, WT GCGR-EGFP, and P86S-EGFP. The apoptotic rates of cells expressing P86S were consistently higher than those of cells expressing EGFP or WT GCGR, starting 2 days after transient transfection (fig. 3A). The difference in apoptotic rates between cells expressing P86S and WT GCGR was largest 4 days after transfection (18.3±0.2% vs 2.1±0.2%, P<0.05). As P86S was significantly localized to endoplasmic reticulum (ER) (data not shown for cells transiently expressing P86S), we tested cleavage of XBP-1 mRNA, a marker of ER stress. Treatment with thapsigargin, a known inducer of ER stress, clearly showed XBP-1 cleavage, but no such cleavage was seen in cells expressing any of the 3 constructs (fig. 3B), suggesting that P86S-induced apoptosis was caused by mechanisms other than ER stress.
 Apoptosis of cells transiently expressing GCGR. H1299 cells were transfected with plasmids encoding EGFP, WT GCGR-EGFP or P86S-EGFP. Cells were replated onto coverslips 1 day after transfection and fixed 2-5 days after transfection. Apoptosis was measured by counting cells with condensed and fragmented nuclei. B) Cleavage of XBP1 mRNA. Untransfected cells treated with 1μM thapsigargin (Sigma, St Louis, IL) for 4hours (Lane 1) or without (Lane 2). H1299 cells in 60-mm dishes were transfected with plasmids encoding EGFP (Lane 5), WT GCGR-EGFP (Lane 4) or P86S-EGFP (Lane 3). Cells were plated into 10-cm dishes 1 day after transfection and total RNA extracted 1 day later. Total RNA was reverse transcribed and XBP1 amplified with PCR. P86S-induced apoptosis was not due to ER stress. Upper band, actin B (233bp); middle band, uncleaved XBP1 (195bp); and lower band, cleaved XBP1 (169bp). EGFP: enhanced green fluorescent protein; GCGR: glucagon receptor; XBP1: X-box binding protein 1.")
Cell apoptosis by WT GCGR and P86S. A) Apoptosis of cells transiently expressing GCGR. H1299 cells were transfected with plasmids encoding EGFP, WT GCGR-EGFP or P86S-EGFP. Cells were replated onto coverslips 1 day after transfection and fixed 2-5 days after transfection. Apoptosis was measured by counting cells with condensed and fragmented nuclei. B) Cleavage of XBP1 mRNA. Untransfected cells treated with 1μM thapsigargin (Sigma, St Louis, IL) for 4hours (Lane 1) or without (Lane 2). H1299 cells in 60-mm dishes were transfected with plasmids encoding EGFP (Lane 5), WT GCGR-EGFP (Lane 4) or P86S-EGFP (Lane 3). Cells were plated into 10-cm dishes 1 day after transfection and total RNA extracted 1 day later. Total RNA was reverse transcribed and XBP1 amplified with PCR. P86S-induced apoptosis was not due to ER stress. Upper band, actin B (233bp); middle band, uncleaved XBP1 (195bp); and lower band, cleaved XBP1 (169bp).
EGFP: enhanced green fluorescent protein; GCGR: glucagon receptor; XBP1: X-box binding protein 1.
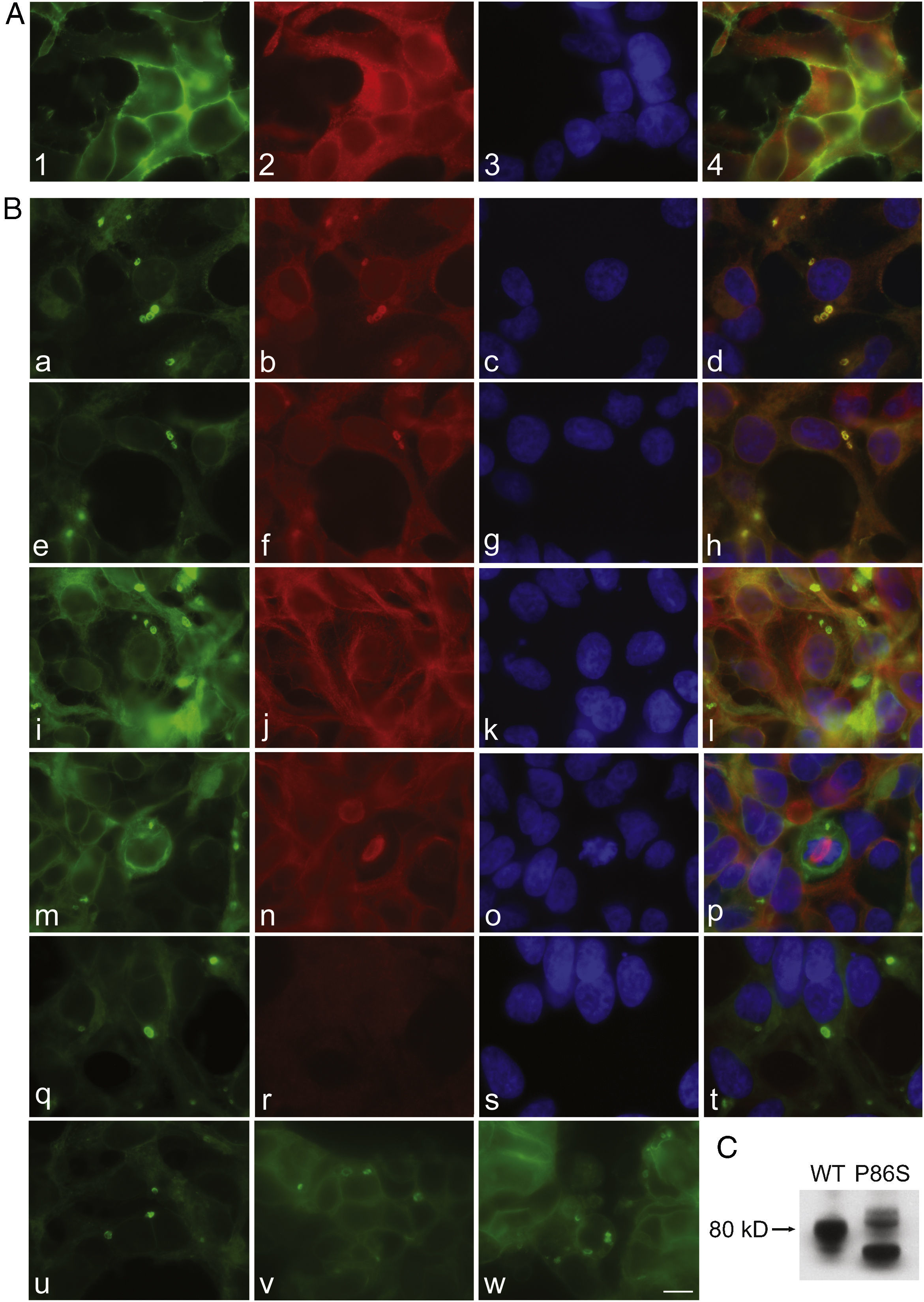
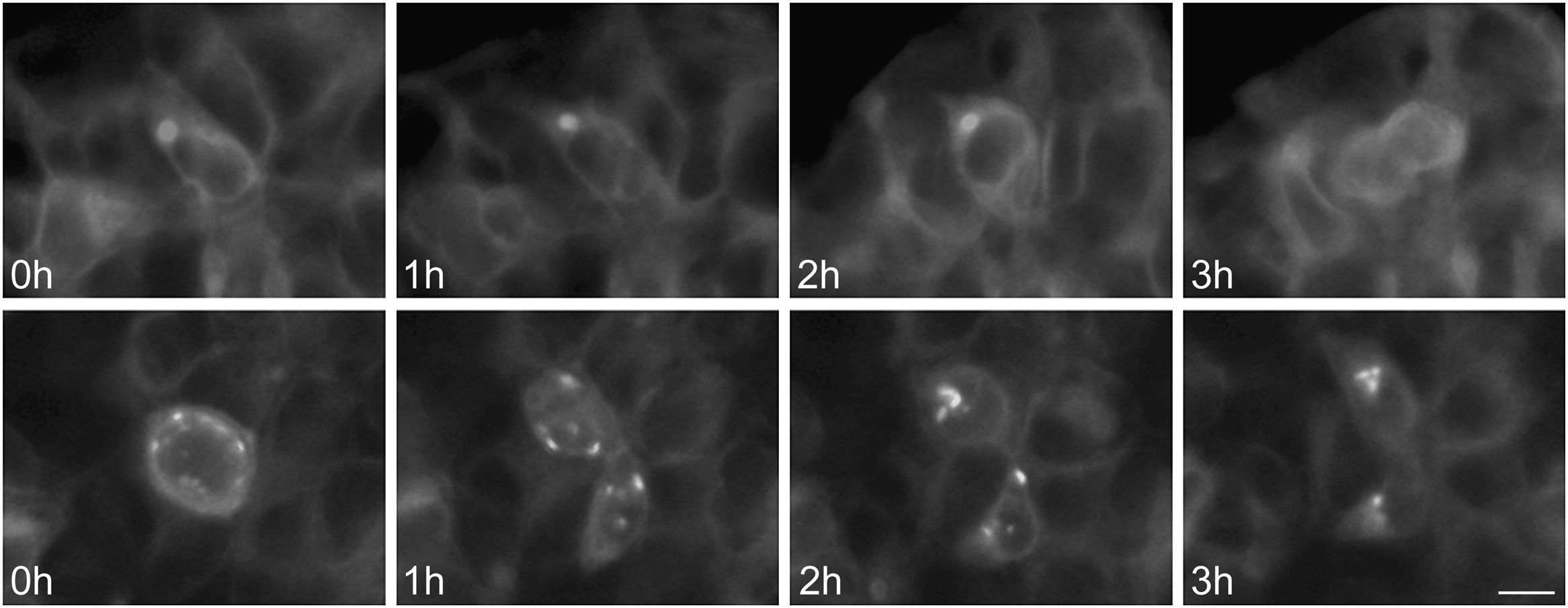
To further examine the effects of P86S expression on cell survival, we established stable HEK 293 cells expressing WT GCGR-EGFP or P86S-EGFP. Apoptotic rates were similar in cells expressing the two constructs: 1.2±0.1% for WT GCGR and 0.9±0.4% for P86S. The expression levels of WT GCGR and P86S were similar (fig. 4). Unlike in cells transiently transfected,11 P86S was processed slightly differently than WT GCGR and appeared as 2 bands close to 80kD (fig. 4C). A circular, inclusion body-like structure labeled by P86S was present in cells stably expressing P86S (24.9±1.9%) but not in those expressing WT GCGR (0±0%, P<0.001) (fig. 4A and B). These novel structures measured 2-3μm in diameter, and were mostly perinuclear and present in all 7 stable clones of HEK 293 cells expressing P86S-EGFP but rarely seen in cells transiently expressing it. Of 70 cells that contain that novel structure, 52 (74%) had 1, 10 (14%) had 2, 5 (7%) had 3, while 3 (4%) had 4 or more. In WT GCGR-expressing cells, WT GCGR and ER marker calregulin were localized distinctly without overlap (fig. 4A). In P86S-expressing cells, P86S and calregulin and glucosidase II, another ER marker, were almost completely colocalized (fig. 4B a-h). The circular, inclusion body-like structures containing P86S were perfectly colocalized with both ER markers. We hereafter call these novel structures circular ER bodies.
 bodies induced by P86S. HEK 293 cells stably expressing WT GCGR-EGFP (Panel A) or P86S-EGFP (Panel B) were fixed and immunofluorescently stained with antibodies against calregulin (2, b), glucosidase II (f), α tubulin (j, n), or ubiquitin (r). The images of WT GCGR-EGFP (1) or P86S-EGFP (a, e, i, m, and q, in green), various cellular markers (2, b, f, n, and r, in red), and nuclei (3, c, g, k, o, and s, in blue) were overlaid (4, d, h, l, p, and t). Yellow or orange shows colocalization of green and red fluorescence. HEK 293 cells stably expressing P86S-EGFP were also treated with proteasome inhibitor LLnL (50μM) (u), microtubule depolymerizer colcemid (100 ng/ml) (v), or both (w), for 8hours. C) Differential processing of WT GCGR-EGFP and P86S-EGFP. HEK 293 cells stably expressing WT GCGR-EGFP and P86S-EGFP were lysed and subjected to western blot using antibody against EGFP. Time of image acquisition is indicated at the left low corner. These results demonstrated that the circular ER bodies are not aggresomes. Bar, 10μm. EGFP: enhanced green fluorescent protein; GCGR: glucagon receptor.")
Characterization of endoplasmic reticulum (ER) bodies induced by P86S. HEK 293 cells stably expressing WT GCGR-EGFP (Panel A) or P86S-EGFP (Panel B) were fixed and immunofluorescently stained with antibodies against calregulin (2, b), glucosidase II (f), α tubulin (j, n), or ubiquitin (r). The images of WT GCGR-EGFP (1) or P86S-EGFP (a, e, i, m, and q, in green), various cellular markers (2, b, f, n, and r, in red), and nuclei (3, c, g, k, o, and s, in blue) were overlaid (4, d, h, l, p, and t). Yellow or orange shows colocalization of green and red fluorescence. HEK 293 cells stably expressing P86S-EGFP were also treated with proteasome inhibitor LLnL (50μM) (u), microtubule depolymerizer colcemid (100 ng/ml) (v), or both (w), for 8hours. C) Differential processing of WT GCGR-EGFP and P86S-EGFP. HEK 293 cells stably expressing WT GCGR-EGFP and P86S-EGFP were lysed and subjected to western blot using antibody against EGFP. Time of image acquisition is indicated at the left low corner. These results demonstrated that the circular ER bodies are not aggresomes. Bar, 10μm.
EGFP: enhanced green fluorescent protein; GCGR: glucagon receptor.
As mutant G protein-coupled receptors such as mutant rhodopsin can localize to a structure called aggresome,19–22 we examined whether the circular ER bodies are aggresomes. Aggresome formation is microtubule-dependent and increases with proteasome inhibition; aggresomes are localized around microtubule organizing centers and contain ubiquitinated proteins. In both interphase (fig. 4B i-l) and metaphase (fig. 4B m-p) cells, the circular ER bodies were distinct from the microtubule organizing centers and they did not colocalize. The circular ER bodies did not colocalize with ubiquitin (fig. 4B q-t). Treatment with proteasome inhibitor LLnL or microtubule depolymerizer colcemid or both did not increase the number or size of the circular ER bodies (fig. 4B u-w). Our results thus demonstrated that the circular ER bodies are not aggresomes.
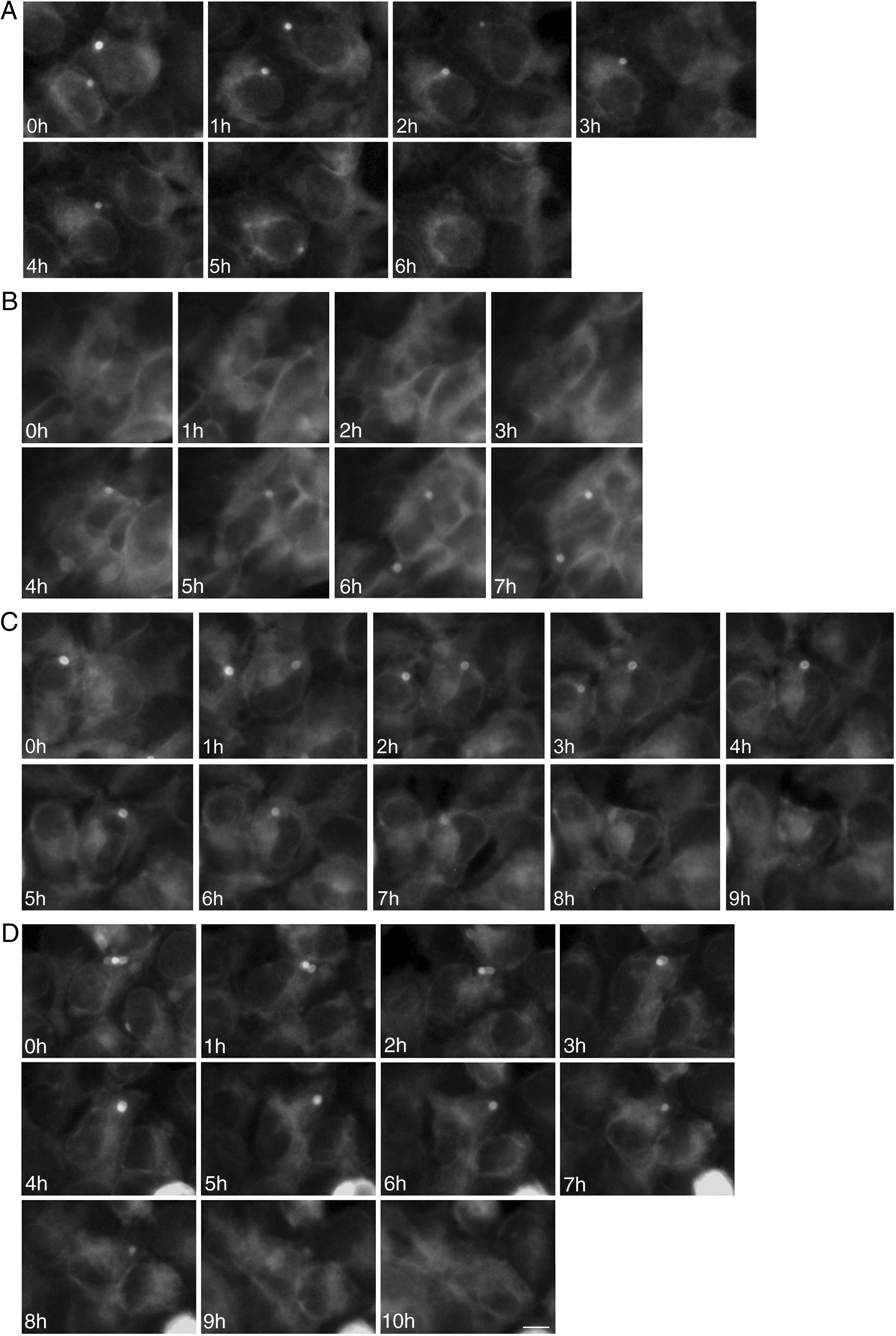
To study the fates of the circular ER bodies and the cells harboring them, we followed 791 live cells over 9-13hours. None of the cells harboring the circular ER bodies died over the observational period and all cells divided normally (fig. 5). The majority of the circular ER bodies disappeared during the observational period (fig. 6A) and new ones emerged with a half-life of 4.1hours (fig. 6B). We observed the entire life cycle of the circular ER bodies in few cells (fig. 6C). Some ER bodies split and fused (fig. 6D).
 bodies recorded by live cell imaging. Live HEK 293 cells stably expressing P86S-EGFP were observed hourly and the same groups of cells were followed over time. Shown are two cells (upper and lower panels) that underwent normal cell division. Time of image acquisition is indicated at the left low corner. Bar, 10μm. EGFP: enhanced green fluorescent protein.")
Division of cells harboring dynamic circular endoplasmic reticulum (ER) bodies recorded by live cell imaging. Live HEK 293 cells stably expressing P86S-EGFP were observed hourly and the same groups of cells were followed over time. Shown are two cells (upper and lower panels) that underwent normal cell division. Time of image acquisition is indicated at the left low corner. Bar, 10μm.
EGFP: enhanced green fluorescent protein.
 bodies revealed by live cell imaging. Live HEK 293 cells stably expressing P86S-EGFP were observed hourly and the same groups of cells were followed over time. A) Disappearance of circular ER bodies from 2 cells, 1 in 3hours, the other in 5hours. B) Emergence of new ER bodies in 2 cells. C) The life cycle of one circular ER body (in the cell in the center of field). Note that an adjacent cell on the left has a dynamic circular EB body that disappeared in 4hours. D) Fusion and disappearance of 3 ER bodies. Time of image acquisition is indicated at the left low corner. The circular ER bodies were dynamic structures. Bar, 10μm. EGFP: enhanced green fluorescent protein.")
Dynamic circular endoplasmic reticulum (ER) bodies revealed by live cell imaging. Live HEK 293 cells stably expressing P86S-EGFP were observed hourly and the same groups of cells were followed over time. A) Disappearance of circular ER bodies from 2 cells, 1 in 3hours, the other in 5hours. B) Emergence of new ER bodies in 2 cells. C) The life cycle of one circular ER body (in the cell in the center of field). Note that an adjacent cell on the left has a dynamic circular EB body that disappeared in 4hours. D) Fusion and disappearance of 3 ER bodies. Time of image acquisition is indicated at the left low corner. The circular ER bodies were dynamic structures. Bar, 10μm.
EGFP: enhanced green fluorescent protein.
The P86S mutant is a naturally-occurring, inactive mutant human GCGR which causes a novel human disease (Mahvash disease) when expressed homozygously.10,11 The Mahvash disease is characterized by hyperglucagonemia without glucagonoma syndrome, hypoglycemia, pancreatic α cell hyperplasia, and pancreatic neuroendocrine tumors. This phenotype is almost identical to that of mice with GCGR deletion and several other similar cases reported previously.14,15,23 Our previous studies demonstrate that P86S exhibits partially defective trafficking and decreased glucagon binding, and requires higher glucagon concentrations for normal cAMP production, establishing that P86S is an inactive mutant but it still has residual signaling through Gs. The previous studies suggest that P86S may exhibit other abnormalities as well. To understand the pathogenesis of the Mahvash disease, we have used transiently or stably expressed EGFP-tagged WT and P86S human GCGR to further study their differences.
The current data clearly demonstrated that unlike the WT GCGR which internalizes upon glucagon binding, the P86S does not internalize even after incubation in very high concentrations of glucagon for an extended period of time. The WT GCGR internalizes with Gsα and GCGR-Gs signal transduction may continue after GCGR internalization at the endo-lysosomal apparatus.8 It appears that besides defective glucagon binding and abnormal receptor localization, failure of P86S internalization may also cause decreased cAMP production.
It is interesting to note that P86S does not mobilize calcium as we have shown previously that high concentrations of glucagon (1μM) produce maximal cAMP signals in cells expressing P86S.11 GCGR increases intracellular calcium levels by releasing calcium from intracellular stores through activating phospholipase C.6,7 It has been suggested that that the increase in intracellular calcium stimulates gluconeogenesis by upregulating the Krebs cycle and the electron-transfer chain and by activating gluconeogenic enzymes.24 As gluconeogenesis is a main function of GCGR in vivo, the absence of calcium signaling by P86S would contribute to decreased neogenesis, explaining in part the hypoglycemia in the patient. How a point mutation in the extracellular domain of GCGR abrogates the coupling between GCGR and Gq is not clear.
Our study also unravels the deleterious effects of transiently expressed P86S on cell survival, adding one more abnormality to P86S. As P86S is significantly localized to the ER and may cause ER stress, we tested cleavage of XBP-1 mRNA, a marker of ER stress,17 induced by P86S expression. Since ER stress is not detected, other unknown mechanisms should underlie the apoptosis induced by transiently expressed P86S. In contrast to cells transiently expressing P86S, those stably expressing P86S do not exhibit excess apoptosis, consistent with the normal liver function of the patient harboring homozygous P86S mutation.10 The cells stably expressing P86S must develop mechanisms to alleviate the toxic effects of P86S, one of which may be the novel, dynamic inclusion body-like structures we termed «circular ER bodies».
Our study has clearly demonstrated that the circular ER bodies are not aggresomes (solid cytoplasmic inclusion bodies19–22) because the former did not change with proteasome inhibition or microtubule disruption, exhibited circular configurations, and were localized away from the microtubule organizing centers. Morphologically the circular ER bodies resemble another inclusion body-like structure called “concentric membranous bodies of the ER” formed by mutant CFTR (cystic fibrosis transmembrane conductance regulator) or wildtype D1 and D2 dopamine receptors, both G protein-coupled,20,25,26 in response to cellular stress. The formation of concentric membranous bodies by mutant CFTR and D1 and D2 receptors requires overexpression of calnexin, an ER chaperone that interacts with glycosylated proteins, and intact microbubules.20,25 In contrast, the circular ER bodies formed by P86S as shown in this study are independent of calnexin overexpression or microtubules, indicating they are different structures from the concentric membranous bodies of the ER. The functions of the circular ER bodies are still unclear but could be inferred from this study as an adaptive response to P86S expression. The circular ER bodies are only present in the stable clones of cells expressing P86S but not in cells transient expressing it, suggesting that they are required for the stable P86S expression. The absence of significant apoptosis in cells stably expressing P86S also supports the notion that the circular ER bodies are protective against the P86S toxicity. We do not have access to the liver or kidney of the patient with Mahvash disease therefore cannot ascertain whether the circular ER bodies are also present in these organs. The circular ER bodies are unlikely an artifact of overexpression as the WT GCGR, expressed at similar levels, did not form them.
In summary, we have described further differences between WT GCGR and P86S in glucagon-induced receptor internalization, calcium mobilization, and effects on cell survival. The P86S mutant induces the formation of dynamic circular ER bodies, which may be adaptive in nature to nullify the P86S toxicity to cells. These differences provide further insights into the pathogenetic mechanisms of the Mahvash disease.
FundingSupported by NIH grant DK071870 (R.Y.) and Cedars-Sinai Medical Center.
Conflict of interestThe authors declare that they have no conflict of interest.
