


El complejo de Carney (CC) es un síndrome descrito por primera vez en 1985, caracterizado por la presencia de mixomas, hiperpigmentación cutánea e hiperactividad endocrina. Es una enfermedad rara, de herencia autosómica dominante con expresividad variable de la que se han descrito unos 600 casos en el mundo. Si bien se han relacionado 2 loci genéticos diferentes, en el 70% de los pacientes se encuentran mutaciones inactivantes del gen PRKAR1A. En un 30% de los pacientes estas mutaciones se producen de novo1.
A continuación presentamos el caso de una paciente con acromegalia en el contexto de un CC.
Se trata de una mujer de 55 años de edad que ingresó en el servicio de Cardiología por un mixoma auricular izquierdo y otro probable en el lado derecho. Durante el ingreso presentó una crisis epiléptica motivo por el que se le solicitó una resonancia magnética nuclear (RNM) craneal que detectó la existencia de una tumoración hipofisaria de 1,2 cm, que cursaba con desplazamiento del tallo hipofisario hacia la derecha, sin compromiso de la región supraselar ni contacto con la región quiasmática.
Entre los antecedentes personales destacar que a los 31 años de edad se le había detectado un mixoma auricular izquierdo, complicado con varios ictus embólicos. Fue resecado y 3 años después recidivó, siendo reintervenido. Como secuela le quedó una epilepsia focal. A los 39 años se le había detectado un carcinoma de mama izquierda. Con 45 años fue valorada en Dermatología por presentar mixomas cutáneos, nevus y lentiginosis desde la adolescencia. A los 54 años se le resecaron varios fibroadenomas en la mama derecha, momento en el que dada la sospecha clínica de un CC se le solicitó estudio genético, sin disponerse del resultado durante la valoración inicial por parte de Endocrinología.
En la anamnesis dirigida, refería notable crecimiento de manos y pies de larga evolución, con aumento del número de zapato del 37 (con 25 años) al 41 actual y dolores articulares de predominio en articulaciones del carpo. No había notado cambios en la voz ni presentaba hipertensión arterial ni alteración del metabolismo hidrocarbonado. Refería aumento de vello solo a nivel de cuero cabelludo. Había presentado ritmo menstrual regular hasta un año antes de la consulta y no refería galactorrea. En la exploración física destacaban estigmas faciales acromegaloides, con macroglosia y marcado engrosamiento de partes blandas. También presentaba lentiginosis en cuello y espalda, así como mixomas cutáneos cervicales.
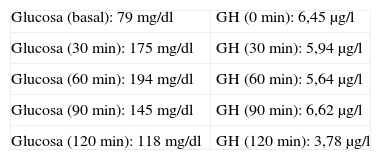
El estudio analítico basal era: tirotropina (TSH) 2,99μUI/ml (0,4-4), tiroxina (T4l) 0,759 ng/dl (0,8-1,9), folitropina (FSH) 90,1 UI/l (posmenopausia 21,7-153), lutropina (LH) 35,6 UI/l (posmenopausia 11,3-39,8), factor de crecimiento insulinoide (IGF-1) 258μg/l (81-225), somatotropina (GH) 5,75μg/l (0,06-5), corticotropina (ACTH) 21,3μg/l (10-46), cortisol 12,9μg/dl (5-25), prolactina (PRL) 9,95μg/l (4,6-37). Se realizó sobrecarga oral de glucosa (SOG) con 75g (tabla 1).
Con el diagnóstico de acromegalia por macroadenoma hipofisario se completó el estudio con una colonoscopia, que detectó varios pólipos adenomatosos túbulo-vellosos con focos de adenocarcinoma en 2 de ellos, y varios adenomas serrados. Se practicó inicialmente una resección de sigma por vía laparoscópica y posteriormente se intervino del macroadenoma hipofisario por vía transesfenoidal.
La anatomía patológica mostraba la mayor parte de la muestra como hipófisis normal y un tercio correspondía a una tumoración homogénea cuyas células teñían exclusivamente para GH.
En la RNM posquirúrgica no se apreciaba imagen de resto tumoral. No obstante persistían unos niveles de IGF-1 aumentados (416) y una SOG patológica, por lo que inició tratamiento con análogos de somatostatina. En siguientes revisiones los niveles de IGF-1 se normalizaron. El despistaje de hipercortisolismo resultó negativo y en la ecografía tiroidea se detectaron varios nódulos infracentimétricos sin datos radiológicos de sospecha.
PRKAR1A es un componente clave de la vía celular de señalización del AMPc implicado en la tumorogénesis. El CC podría ser considerado una forma de neoplasia endocrina múltiple con afectación de glándulas suprarrenales, hipófisis, tiroides y gónadas.
El diagnóstico clínico se realiza por la presencia de 2 o más manifestaciones típicas confirmadas por histología, pruebas de laboratorio o de imagen. Estas manifestaciones incluyen lentiginosis cutánea con distribución típica (labios, conjuntiva, mucosa oral y genital), nevus azul, schwanoma melanocítico psamomatoso, mixomas cutáneos o mucosos, mamarios o cardíacos, hiperplasia adrenal micronodular pigmentada (PPNAD), acromegalia, carcinoma de tiroides o presencia de múltiples nódulos hipoecoicos, tumor de células grandes calcificadas de Sertoli, osteocondromixoma y/o múltiples adenomas mamarios ductales. Si el paciente es portador conocido de una mutación o familiar de primer grado de un afectado, únicamente requiere una manifestación para ser diagnosticado2. El estudio genético debería ser propuesto a todos los casos índice y si se detecta mutación también a los familiares de primer grado3. La manifestación endocrinológica más frecuente y específica es la PPNAD. Los mixomas cardíacos son la causa más común de mortalidad, bien de forma directa o derivada de complicaciones quirúrgicas. Pueden ser múltiples, aparecer a edades más tempranas y presentan mayor riesgo de recurrencia que los esporádicos4,5.
En la descripción inicial de 1985, 4 de 40 pacientes con CC presentaban adenomas productores de GH4. Series más recientes sitúan la prevalencia en un 12%. Sin embargo, un porcentaje mayor de pacientes presentan alteraciones del eje en forma de elevación basal de GH/IGF-1 o ausencia de frenado en la SOG6,7. La acromegalia en el CC presenta un curso lento e insidioso. La edad media de presentación es de 35 años y generalmente se presenta como un macroadenoma1.
A diferencia de los pacientes con CC, los ratones heterocigotos para mutaciones en PRKAR1A no desarrollan tumores hipofisarios, aunque en ratones knockout para este gen en el linaje Pit1 hipofisario sí se producen adenomas secretores de GH. En base a estos modelos animales, se ha postulado que se precisa una pérdida completa del alelo sano para el desarrollo de adenomas productores de GH en el CC. No se han detectado mutaciones en el gen PRKAR1A en casos esporádicos, pero algunos datos sugieren que mecanismos no genómicos pudieran causar una pérdida de la expresión de esta proteína que contribuyera a su desarrollo8.
A nivel histopatológico, los adenomas en el CC pueden ser similares a los esporádicos, presentarse de forma multifocal o con focos de hiperplasia de células somatotropas. La presencia de hiperplasia generalizada de células somatotropas en las autopsias de pacientes con CC ofrece una explicación a la elevada prevalencia de alteraciones bioquímicas del eje somatotropo en estos pacientes9. A diferencia de nuestro caso, la mayoría tiñen para varias hormonas, fundamentalmente para PRL, pero también es posible la tinción para subunidad-α, TSH-β, LH-β y ocasionalmente FSH-β.
El 72% de los pacientes presentan simultáneamente acromegalia y PPNAD9. En nuestro caso, no se ha detectado PPNAD.
Nuestra paciente cumplía criterios clínicos diagnósticos y posteriormente el estudio genético confirmó la mutación en el gen PRKAR1A. Actualmente sus hijos no cumplen criterios clínicos diagnósticos y se encuentran pendientes del estudio genético.
