


The autoimmune polyglandular syndromes are infrequent associations of endocrine disorders characterized by the coexistence of at least two glandular insufficiencies due to autoimmune mechanisms, with the possible association of other non-endocrine autoimmune diseases. Autoimmune chronic urticaria (ACU) has been associated with Hashimoto's thyroiditis. To date, the literature has not described ACU as a component of autoimmune polyglandular syndrome (APS).
A 33-year-old woman presented with the following family history: father with Hashimoto's thyroiditis and a female sibling with vitiligo and alopecia areata. The patient had been followed-up in the endocrinology clinic since the age of 14 years, when type 1 diabetes mellitus was diagnosed, initially manifesting with cardinal symptoms and ketosis. At 16 years of age the patient visited the emergency service with asthenia and progressive pigmentation of the lips and knuckles. Hyponatremia and hyperpotassemia were detected. The low cortisol and aldosterone levels and ACTH elevation confirmed the diagnosis of Addison's disease.
At 20 years of age she presented increased stool frequency, fine tremor and thinning, together with tachypsychia and tiredness. The thyroid gland functional study revealed total T4>6ng/dl; T3: 375ng/ml; TSH<1mU/l; antiperoxidase antibodies (anti-TPO): 518U/ml (50–75); antithyroglobulin antibodies (anti-TG): 550IU/ml (100–150); and anti-TSH receptor antibodies (TRAb): 11.6IU/l (0–2). Thyroid scintigraphy confirmed diffusely increased uptake consistent with Graves-Basedow's disease. After a year and a half of treatment with synthetic antithyroid drugs, and due to the persistence of hyperthyroidism, I-131 therapy was indicated, which produced iatrogenic hypothyroidism. Since then the patient has been receiving replacement therapy. The autoantibody titers showed gradual negative conversion to anti-TPO 31.2IU/ml and anti-TG 119.7IU/ml.
Six years later the patient developed recurrent, evanescent pruriginous erythematous wheal-like lesions, associated with several episodes of glottic edema. Treatment was provided with different combinations of antihistamines and corticosteroids, though without success. The skin biopsy findings were compatible with urticaria. Immunoglobulin tests were normal, with the exception of IgA deficiency. Alpha-trypsin and complement factors C3 and C4, as well as factor B, were normal. Serological testing for HBV, HCV and HIV proved negative. With the suspicion of autoimmune chronic urticaria (ACU), autologous serum testing was carried out and proved positive, thereby confirming the disease. Following the failure of other treatments with oral antihistamines and corticosteroids, cyclosporine was started, followed by control of the urticaria.
Coinciding with the development of urticaria, the patient showed an increase in antithyroid antibodies (anti-TPO: 139.1IU/ml; anti-TG: 409.7IU/ml; TRAb: 3.75IU/l), with a posterior decrease in their respective titers – though this was not associated with improvement of the skin lesions. At the time of writing the patient is receiving immunosuppressive therapy, and is unable to tolerate suspension of the latter, due to reappearance of the lesions.
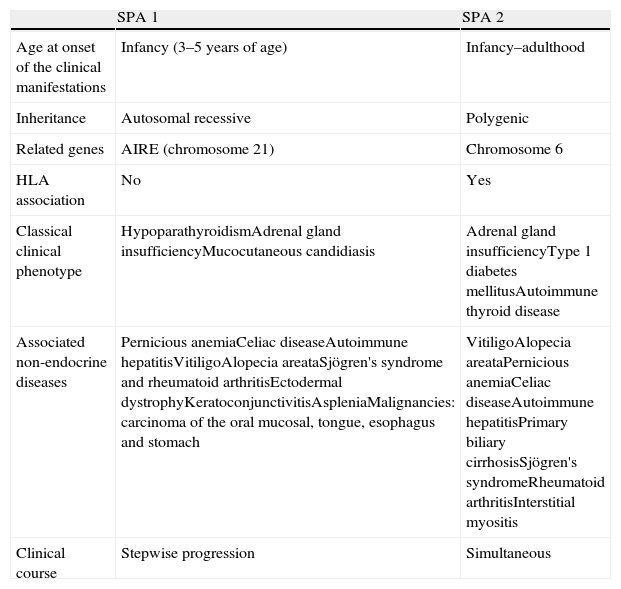
The autoimmune polyglandular syndromes are characterized by alterations of at least two endocrine glands due to autoimmune mechanisms, with the possible association of other non-endocrine autoimmune diseases. There are two main types (Table 1). APS 2, with an incidence of 1.5–4.5 cases per 100,000 inhabitants/year, is more common that APS 1.1 The most frequent endocrine alterations of this syndrome are autoimmune thyroid disease (with a similar prevalence of Graves disease and Hashimoto's thyroiditis), type 1 diabetes mellitus and Addison's disease.2 The most common situation is the presence of only two of these components in the course of the patient's life. A triple association of these diseases is infrequent, being observed in only 10–12% of the cases.1
Characteristics of the main types of autoimmune polyglandular syndrome.
| SPA 1 | SPA 2 | |
| Age at onset of the clinical manifestations | Infancy (3–5 years of age) | Infancy–adulthood |
| Inheritance | Autosomal recessive | Polygenic |
| Related genes | AIRE (chromosome 21) | Chromosome 6 |
| HLA association | No | Yes |
| Classical clinical phenotype | HypoparathyroidismAdrenal gland insufficiencyMucocutaneous candidiasis | Adrenal gland insufficiencyType 1 diabetes mellitusAutoimmune thyroid disease |
| Associated non-endocrine diseases | Pernicious anemiaCeliac diseaseAutoimmune hepatitisVitiligoAlopecia areataSjögren's syndrome and rheumatoid arthritisEctodermal dystrophyKeratoconjunctivitisAspleniaMalignancies: carcinoma of the oral mucosal, tongue, esophagus and stomach | VitiligoAlopecia areataPernicious anemiaCeliac diseaseAutoimmune hepatitisPrimary biliary cirrhosisSjögren's syndromeRheumatoid arthritisInterstitial myositis |
| Clinical course | Stepwise progression | Simultaneous |
The non-endocrine autoimmune diseases most commonly associated with APS 2 are vitiligo (19.9%), alopecia areata (9.6%) and pernicious anemia (5.3%).2
Our patient presented three endocrine disorders in a stepwise manner, though in many cases of APS 2 these diseases can develop simultaneously.
Autoimmune chronic urticaria (ACU) is characterized by recurrent, evanescent pruriginous erythematous wheal-like lesions, with a duration of at least 6 weeks. The disorder is often associated with angioedema, which develops when the deep dermis and subcutaneous cellular tissue are affected.3 The origin of the disease is not known. In a large percentage of cases no etiological diagnosis is established, these presentations being described as idiopathic chronic urticaria. In recent years it has been shown that 45% of all cases of chronic urticaria appear to have an autoimmune origin.4 The presence of autoantibodies activates the basophils and mast cells, leading to degranulation and the release of histamine, among other cytokines.5 The biopsy in turn shows tissue edema, vascular dilatation, mast cell degranulation and a perivascular infiltration composed of CD4+/CD8+lymphocytes, eosinophils, basophils and neutrophils.6
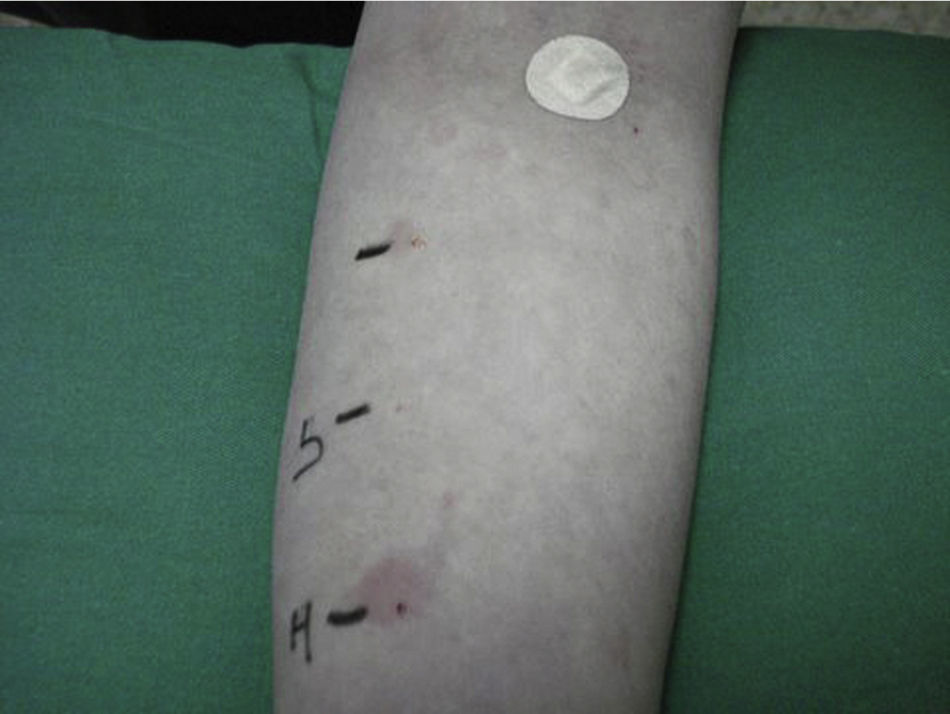
The mean duration of the disease is 3–5 years, and in 20% of the cases it persists for more than 5 years. The factors associated with a longer duration of the illness include the presence of angioedema and autoimmunity.6 Autologous testing is used to diagnose autoimmune chronic urticaria, involving the intradermal injection of autologous serum into the skin of the patient, and using physiological saline as a negative control and histamine as a positive control. The result is considered to be pathological when a wheal 1.5mm larger than the saline control is induced (Fig. 1). The presence of other autoantibodies, especially antiperoxidase antibodies, indirectly supports the diagnosis of the disease. The prevalence of these antibodies in autoimmune chronic urticaria is 10–29%7; in contrast, there are very few references regarding its association with Graves-Basedow's disease, or the presence of anti-TSH receptor antibodies.8 We have found no mention in the literature of the presence of ACU as a component of APS, though in some cases it has been found to be associated with other autoimmune diseases such as celiac disease, type 1 diabetes mellitus, juvenile chronic arthritis, and within families with autoimmune diseases.9 In our case, ACU could have developed as a non-endocrine component of APS with an antecedent of Graves-Basedow's disease, instead of the most common antecedents (usually Hashimoto's thyroiditis). The patient moreover had a family history of autoimmune diseases.

Regarding the time of appearance, ACU can manifest before, during or after the onset of thyroiditis.9 Some studies have described an improvement of urticaria upon the normalization of thyroid function, despite the persistence of antibodies,10 though other studies have found no such association.9 Likewise, no correlation has been observed between clinical resolution and antibody levels.11 In our case, the appearance of ACU showed a time relationship with antithyroid antibody elevation, though the urticaria persisted despite a reduction in antibody titer after the start of immunosuppressive therapy.
In conclusion, the interest of this clinical case is the possible albeit unusual association of APS 2 with its three classical components, and with autoimmune chronic urticaria. This association, and the family antecedents identified in our patient, together with the associations described in other recent publications involving other autoimmune diseases, could define chronic urticaria as another manifestation of a broad autoimmune spectrum – in addition to its already known association with Hashimoto's thyroiditis.
Conflicts of interestThe authors state that they have no conflicts of interest.
Please cite this article as: Ramos-Prol A, et al. Urticaria crónica autoinmune como posible manifestación no endocrina de un síndrome poliglandular autoinmune tipo 2. Endocrinol Nutr. 2011;58(9):497–505.
