


Multiple endocrine neoplasia type 2 (MEN 2A) syndrome is clinically characterized by a tendency to develop medullary thyroid carcinoma, pheochromocytoma, and primary hyperparathyroidism. Any association with other types of tumor is extremely rare. A case of glioblastoma multiforme1 and two cases of adrenal ganglioneuroma2 have been reported in patients with MEN 2. A case of gastrointestinal stromal tumor in an adult with MEN 2A3 and three cases of head and neck paraganglioma in patients with MEN 2A have also been reported.4,5 The development of a metastatic alveolar rhabdomyosarcoma in a child with MEN 2A has also recently been reported.6 Finally, there have also been reports of some patients with acromegaly and MEN 2. Lipomas and meningiomas have been associated with MEN 1 syndrome, but not with MEN 2.
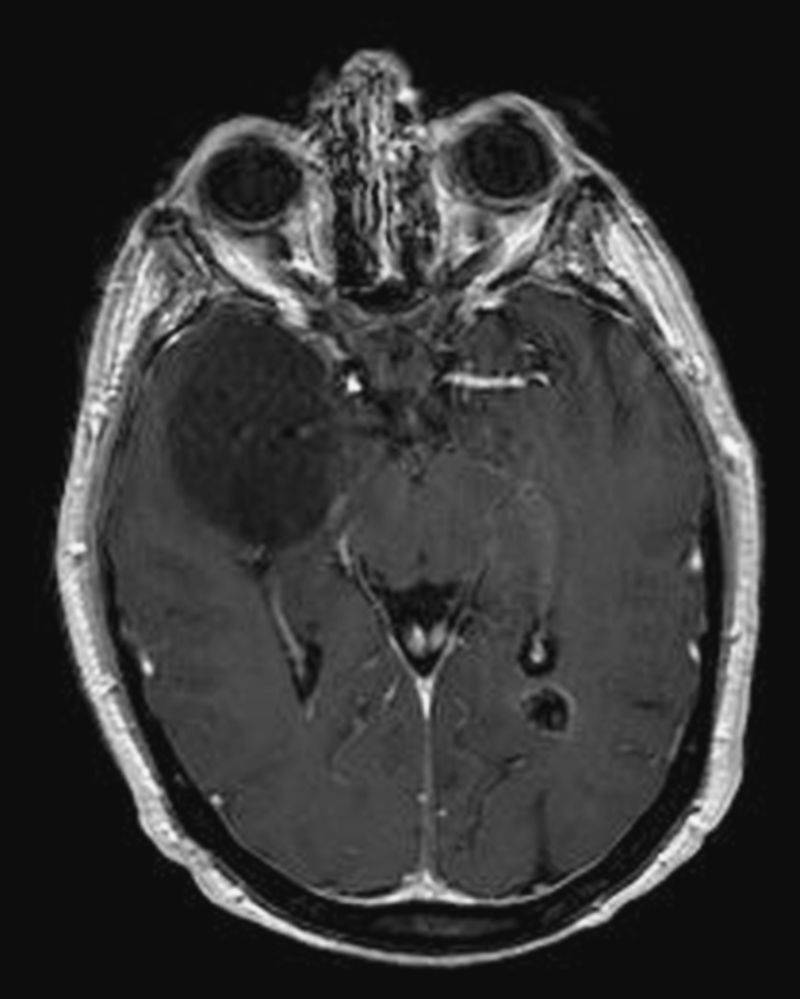
We report the case of a 39-year-old female patient who had undergone total thyroidectomy at 26 years of age when a familial study, performed after her mother was diagnosed with MEN 2A, found a C618R mutation in exon 10 of the RET proto-oncogene. A multifocal medullary thyroid carcinoma was found in the surgical specimen. Seven years later, the patient underwent right laparoscopic adrenalectomy for pheochromocytoma. At 36 years of age, she was diagnosed with a posterior cervical lipoma based on ultrasonographic appearance. The patient remained symptom-free and with no evidence of disease until she attended the emergency room in 2010 for an episode of generalized seizures. Magnetic resonance imaging of the brain revealed a large tumor (54mm×51mm×42mm) in the white matter of the right temporal lobe (Fig. 1). The tumor caused no vasogenic edema and showed no contrast uptake upon gadolinium injection. Incidentally, a 12-mm calcified meningioma was found in the occipital horn of the left lateral ventricle (Fig. 1). A stereotactic biopsy was performed in the temporal tumor, leading to diagnosis of diffuse astrocytoma (grade II glioma according to the classification of the World Health Organization [WHO]). The patient's mother, the only other family member affected, had developed no other tumors apart from medullary thyroid carcinoma and pheochromocytoma.

Axial section of magnetic resonance imaging of the brain showing a large tumor in the white matter of the right temporal lobe. The tumor did not cause vasogenic edema, but induced the partial collapse of the right temporal ventricle. A calcified meningioma in the occipital horn of the left lateral ventricle is also seen.
MEN 2 syndrome is due to the presence of germinal mutations in the RET proto-oncogene. RET is expressed in the central and peripheral nervous system, in cells derived from the neural crest and the urogenital crest. It transduces cell growth and differentiation signals. Normal RET activation depends on the binding of specific ligands belonging to the family of glial cell-derived proteins (GDNF). Mutations associated with MEN 2 syndrome cause receptor molecule dimerization and the activation of ligand-independent intracellular signaling pathways. It has been noted that the phenotype of diseases caused by RET mutations may be modified by the presence of another mutation7 and GDNF,8 that is, other mutations or factors may modify the clinical expression of a syndrome in a particular patient. Although the occurrence of other tumors has only exceptionally been reported in MEN 2A, we think it unlikely that the development of three tumors (lipoma, meningioma, and astrocytoma) in a patient with a hereditary cancer syndrome is due to chance.
Gliomas are primary brain tumors showing histological characteristics of glial cells (astrocytes, oligodendrocytes, and ependymal cells). They are classified according to their differentiation line and histological grade of malignancy. The WHO classifies astrocytomas into four prognostic grades: grade I (pilocytic astrocytoma), grade II (diffuse astrocytoma), grade III (anaplastic astrocytoma), and grade IV (glioblastoma). Low-grade gliomas (grade I and II tumors) have been called “benign”, but are only occasionally associated with long-term survival and frequently develop characteristics similar to those of more aggressive tumors. High-grade or “malignant” gliomas include grade III and IV tumors.
Some data appear to suggest that the RET proto-oncogene may play a role in the development of glial tumors. Glial tumors overexpress GDNF,9 which has been identified as a potent neurotrophic factor for a variety of neuronal cell lines.10,11 Several studies suggest that GDNF may participate in the proliferation of these tumors12,13 and induce cell migration from gliomas, neuroblastic tumors,14–16 and other types of tumor.
To sum up, our patient developed three tumors apparently not associated with MEN 2A. While this may be due to chance, some data suggest that at least the coexistence of glioma and MEN 2A may not be casual. Studies are needed to clarify the potential association between MEN 2 and other types of tumor.
Please cite this article as: Isidro ML, et al. Un caso de astrocitoma en una paciente con síndrome de neoplasia endocrina múltiple tipo 2A. ¿Podría no ser casual la asociación de tumores gliales y síndrome de neoplasia endocrina múltiple 2A? Endocrinol Nutr. 2011;58(9):497–505.
