



El complejo de Carney (CNC) es un síndrome de herencia autosómica dominante caracterizado por mixomas en diferentes localizaciones, hiperpigmentación cutánea y afectación de glándulas endocrinas. La afectación cutánea más característica es la presencia de lentiginosis y nevus azules. Los mixomas pueden hallarse en tejido subcutáneo, mamario o en región cardíaca. Los mixomas cardíacos pueden ser múltiples, localizados en cualquier cámara cardíaca y presentan mayor riesgo de recurrencia. La manifestación endocrinológica más frecuente es la presencia de un síndrome de Cushing independiente de ACTH debido a enfermedad adrenocortical nodular pigmentada primaria (primary pigmented nodular adrenal disease o PPNAD). La PPNAD puede manifestarse de manera aislada sin otras manifestaciones del CNC. Otras glándulas afectadas son la hipófisis, tiroides y gónadas.
El gen PRKAR1A localizado en 17q22-24 codifica la subunidad reguladora R1A de la proteína kinasa A. Mutaciones inactivantes de este gen se encuentran en un 70% de pacientes con CNC. PRKAR1A es un componente clave de la vía celular de señalización del AMPc implicado en la presencia de tumorogénesis de origen endocrinológico. Se han descrito numerosas mutaciones en el gen PRKAR1A y la práctica totalidad de ellas conllevan cambios en la secuencia que dan lugar a codones de terminación prematuros y el RNAm resultante es degradado por la mRNA nonsense mediated decay. No existe una clara relación entre genotipo y fenotipo en los pacientes con CNC.
Se ha de realizar estudio genético en todos los casos índice. El seguimiento para todas las manifestaciones clínicas del CNC deberían realizarse anualmente en los pacientes afectados. El estudio genético posibilita preparar de manera más efectiva nuevas estrategias terapéuticas y consejo genético para los pacientes y portadores, y evita la realización de pruebas innecesarias en familiares no portadores.
Carney complex (CNC) is an autosomal dominantly inherited syndrome characterized by spotty skin pigmentation, cardiac and cutaneous myxoma, and endocrine overactivity. Skin pigmentation includes lentigines and blue nevi. Myxomas may occur in breast, skin and heart. Cardiac myxomas may be multiple and occur in any cardiac chamber, and are more prone to recurrence. The most common endocrine gland manifestation is an ACTH-independent Cushing's syndrome due to primary pigmented nodular adrenocortical disease (PPNAD). PPNAD may occur isolated, with no other signs of CNC. Pituitary and thyroid glands and gonads are also involved.
The PRKAR1A gene, located in 17 q22-24, encodes type 1A regulatory subunit of protein kinase A. Inactivating germline mutations of this gene are found in 70% of patients with CNC. PRKAR1A is a key component of the c-AMP signaling pathway that has been implicated in endocrine tumorigenesis. Many different mutations have been reported in the PRKAR1A gene. In almost all cases the sequence change was predicted to lead to a premature stop codon and the resultant mutant mRNA was subject to nonsense-mediated mRNA decay. There is no clear genotype-phenotype correlation in patients with CNC.
Genetic analysis should be performed in all CNC index cases. All affected patients should be monitored for clinical signs of CNC at least once a year. Genetic diagnosis allows for more effective preparation of more appropriate and effective therapeutic strategies and genetic counseling for patients and gene carriers, and to avoid unnecessary tests to relatives not carrying the gene.
El complejo de Carney (CNC) (MIM:160980) fue descrito por primera vez por J. Aidan Carney et al, en 1985, como una combinación de mixomas, hiperpigmentación cutánea e hiperactividad endocrina. Con la descripción de este nuevo síndrome se constató que la gran mayoría de los pacientes previamente caracterizados bajo entidades diferentes conocidas por los acrónimos NAME (nevi, atrial myxoma, ephelides) y LAMB (lentigines, atrial myxoma, blue nevi) deberían ser incluidos dentro del CNC. El CNC es un síndrome de herencia autosómica dominante que muestra expresividad variable y penetrancia casi completa (70-80% a los 40 años). El CNC podría ser considerado una forma de neoplasia endocrina múltiple por la frecuente afectación de dos o más glándulas endocrinas, incluyendo enfermedad adrenocortical nodular pigmentaria primaria (primary pigmented nodular adrenal disease, PPNAD), adenomas hipofisarios productores de GH y PRL, tumores testiculares, enfermedad nodular de tiroides y quistes ováricos, siendo algunas como la PPNAD altamente específicas de la misma1,2.
En la actualidad, el diagnóstico de este síndrome se realiza por la presentación de dos o más manifestaciones típicas del mismo confirmadas por histología, pruebas de laboratorio o pruebas de imagen. Asimismo, si el paciente es portador conocido de una mutación inactivante de la subunidad reguladora 1 alfa de la proteína kinasa A (PRKAR1A) o es familiar de primer grado de un paciente afectado de CNC únicamente requiere una manifestación clínica para poder ser diagnosticado de la enfermedad3 (tabla 1).
Criterios diagnósticos del complejo de Carney
| - Lentiginosis cutánea con distribución típica (labios, conjuntiva, mucosa oral y genital |
| - Mixoma (cutáneo o mucoso)a |
| - Mixoma cardíaco |
| - Mixomatosis mamariaa o imagen en RMN (modo supresión de grasa) sugestiva de diagnóstico |
| - PPNADa o respuesta paradójica positiva de corticoesteroides en orina tras la administración de dexametasona durante el test de Liddle |
| - Acromegalia secundaria a adenoma productor de GHa |
| - LCCSCTa o presencia de calcificaciones características en ecografía testicular |
| - Carcinoma de tiroidesa o presencia de múltiples nódulos hipoecoicos en ecografía de tiroides en paciente joven |
| - Schwanoma melanocítico psamomatosoa |
| - Nevus azul, múltiples nevus azules epitelioidesa |
| - Múltiples adenomas mamarios ductalesa |
| - Osteocondromixoma |
| Criterios adicionales |
| - Familiar de primer grado afectado |
| - Mutación inactivante del gen PRKAR1A |
Para realizar el diagnóstico de CNC, el paciente ha de presentar: 1) dos o más manifestaciones clínicas de las mencionadas en el listado superior o 2) una manifestación clínica y un criterio adicional (familiar de primer grado afectado o presentar mutación inactivante del gen PRKAR1A). LCCSCT: tumor de células grandes calcificadas de Sertoli (large-cell calcifying Sertoli cell tumor); PPNAD: enfermedad adrenocortical nodular pigmentaria primaria (primary pigmented nodular adrenal disease).
La determinación de mutaciones inactivantes de la subunidad reguladora tipo 1A (R1A) del gen de la proteína kinasa A (PRKAR1A) en la mayoría de los pacientes con CNC han permitido una estimación de su penetrancia, la validación de los criterios diagnósticos de la enfermedad y la constatación de que no existe una clara relación fenotipo-genotipo.
EpidemiologíaEl CNC es una enfermedad extremadamente infrecuente. Se han descrito aproximadamente 600 casos, siendo la serie más larga la presentada por la Clínica Mayo y el Hospital de Cochin en 2009 con 353 pacientes. Los datos que se desprenden de esta serie hablan de una mayor prevalencia en el sexo femenino (63%) y un diagnóstico que se realiza entre la segunda y tercera décadas de la vida. El 73% presentaba mutaciones inactivantes del gen PRKAR1A. Se han descrito casos en todas las etnias4.
Manifestaciones clínicas no endocrinológicasLesiones cutáneasLa manifestación clínica más frecuente en el CNC es la lentiginosis cutánea. El hecho de ser tan característica y generalmente tan precoz la hace muy útil a la hora del diagnóstico. Puede manifestarse desde el nacimiento aunque no adquiere su distribución típica hasta la adolescencia. La lentiginosis tiende a desaparecer a partir de la cuarta década de la vida. Presenta una distribución característica alrededor de labio superior e inferior, conjuntiva y mucosa oral y genital.
Los mixomas cutáneos son pápulas o nódulos subcutáneos de aspecto liso y color anacarado o rosado. Las lesiones son generalmente múltiples y localizadas de manera preferente en el canal auditivo externo, los párpados y la región mamaria.
Es frecuente la presencia de múltiples nevus azules y nevus azules epitelioides.
Mixoma cardíacoEl mixoma cardíaco se trata de la manifestación no cutánea más frecuente. Es responsable de más del 50% de la mortalidad en pacientes con CNC secundaria a fenómenos embólicos, fallo cardíaco u oclusión valvular. Pueden aparecer en cualquier cavidad cardíaca, ser múltiples y presentar una agresividad mayor y una mayor tasa de recurrencia que en las formas esporádicas.
Otros tumoresLa presencia de estos tumores en el CNC son muy infrecuentes, pero también muy específicos. El schwanoma psamomatoso melanocítico se asienta en el sistema nervioso periférico y central con preferencia por el tracto gastrointestinal. Debe su nombre a que presenta hiperpigmentación característica debido al acúmulo de melanina con frecuentes calcificaciones. Otros tumores característicos son los adenomas ductales de mama, mixomas de mama y osteocondromixomas3,5–7.
Manifestaciones endocrinológicasEnfermedad adrenocortical nodular pigmentada primariaManifestaciones clínicasLa PPNAD es una causa muy infrecuente de síndrome de Cushing independiente de ACTH (< 1%). Generalmente se presenta en el contexto del CNC y es el tumor de glándula endocrina más frecuente. La incidencia de PPNAD es del 26% en los pacientes con CNC en el momento del diagnóstico. La PPNAD también puede diagnosticarse sin otras manifestaciones del CNC o historia familiar de la enfermedad (PPNAD aislada). La edad media en el momento del diagnóstico es de 34 años y es significativamente más frecuente en mujeres (71%) y diagnosticado a edades más tempranas (30 años versus 46 años en hombres)4.
Debe su nombre a su aspecto macroscópico, caracterizado por pequeños nódulos hiperpigmentados situados en la corteza suprarrenal de manera bilateral, aunque también se han descrito nódulos de mayor tamaño.
El diagnóstico de síndrome de Cushing en la PPNAD puede ser complicado debido a que el hipercortisolismo puede manifestarse de muy diversas maneras. Puede presentarse como lentamente progresivo, explosivo, de manera cíclica, asintomática (formas subclínicas) y en algunos casos remitir espontáneamente. Las formas paucisintomáticas o asintomáticas suelen ser diagnosticadas al realizarse screening sistemático en los pacientes con PPNAD, ya sea en pacientes con otras manifestaciones clínicas de la enfermedad o en el cribaje a familiares.
A pesar de la variabilidad en la forma de presentación, las manifestaciones clínicas secundarias del hipercortisolismo son similares a las del resto de etiologías causantes de Cushing (obesidad troncular, ganancia ponderal, estrías rojo vinosas, debilidad muscular, osteoporosis y talla baja en niños)3–7.
Estudio hormonalLa PPNAD suele mostrar concentraciones de cortisol libre urinario elevadas en el momento del diagnóstico en la mayoría de pacientes, aunque sus valores pueden ser muy variables. El ritmo circadiano de cortisol está abolido y los niveles de ACTH plasmático suprimidos. En las formas poco sintomáticas, es frecuente la discrepancia analítica entre niveles de ACTH plasmático suprimidos y valores de cortisol plasmático o urinario en niveles dentro del rango normal. No existe estimulación de cortisol o ACTH tras la estimulación con CRH5–8. No se consigue una supresión de los niveles de cortisol plasmático tras frenación con dexametasona incluso a altas dosis. Lo más característico (incluido en los criterios diagnósticos) es que se puede observar un aumento paradójico de los niveles de cortisol libre urinario tras administración de dexametasona a dosis altas durante la realización del test de Liddle de 6 días. El test consiste en determinaciones basales de cortisol libre urinario durante los dos primeros días, los siguientes dos días se realiza determinación de cortisol libre urinario tras administración de dosis bajas de glucocorticoides (0,5 mg/6 h en adultos o 7,2 μg/kg/6 h en niños) y en los días 5 y 6 se realiza la medida tras administración de dosis altas de glucocorticoides (2 mg/6 h en adultos y 28,5 μg/kg en niños). Este test permite distinguir la PPNAD del resto de enfermedades primarias nodulares suprarrenales (adenoma suprarrenal, enfermedad macronodular adrenal). La elevación de los niveles de cortisol urinario > 100% en el día 6 es diagnóstico de PPNAD (61% de falsos negativos), aunque una elevación > 50% ocurría en el 69% de PPNAD, frente a sólo el 20% de adenomas únicos y el 0% de las hiperplasias macronodulares bilaterales. Esta prueba es muy útil para poder diagnosticar de PPNAD al subgrupo de pacientes que presenta discrepancia ente los niveles de ACTH y valores de cortisol libre urinario9.
Estudios posteriores sugieren que esta característica diferenciadora de la PPNAD se debe a un efecto directo de los glucocorticoides sobre las suprarrenales y que éstas presentan un mayor número de receptores de glucocorticoides en sus nódulos que las adrenales normales10,11.
Pruebas de imagenLas glándulas suprarrenales en la PPNAD son normales en tamaño y peso. Por este motivo, las suprarrenales pueden aparecer como normales en tomografía computadorizada (TC) en más de un tercio de los pacientes. En el resto de pacientes se presentan como micronódulos o en algunos casos, incluso, como macronódulos en una o ambas suprarrenales. La RMN no mejora los resultados de la tomografía. Inicialmente, se describió una imagen típica radiológica (bed and string) para la PPNAD que se ha ido abandonando por su falta de especificidad.
La realización de gammagrafía con yodo colesterol también puede ser útil en el diagnóstico pues generalmente muestra una captación bilateral de las suprarrenales. Sin embargo, la falta de comercialización del radiotrazador ha imposibilitado su utilización en el estudio de la PPNAD en los últimos años8.
Anatomía patológicaMacroscópicamente, las suprarrenales presentan alteraciones bilaterales en forma de múltiples nódulos de pequeño tamaño (< 4 mm) de color negro, marrón, verde oscuro o incluso rojo o amarillo, presencia de atrofia cortical y desorganización estructural.
Microscópicamente, los nódulos están formados por células corticales hiperplásicas con citoplasma amplio eosinófilo que contiene lipofucsina. Los nódulos son positivos a la sinaptofisina en la inmunohistoquímica.
Es destacable que en las autopsias realizadas a pacientes con CNC revelaban alteraciones morfológicas características de la PPNAD en las glándulas suprarrenales incluso cuando no presentaban signos de hipercortisolismo. Esto sugiere que la PPNAD es una manifestación constante en el CNC, aunque con variables grados de expresividad clínica12.
AcromegaliaLa presencia de acromegalia clínicamente significativa secundaria a adenomas productores de GH ocurre en un 12% de los pacientes con CNC. La acromegalia en el CNC se caracteriza por un curso lento e insidioso. La edad media de presentación es de 35 años aproximadamente4.
La aparición de acromegalia bioquímica (elevación de GH e IGF-1 con leve hiperprolactinemia) puede presentarse hasta en un 75% de los pacientes con CNC. Generalmente, estas alteraciones bioquímicas son desenmascaradas con la aparición de resultados anormales tras la realización del test de sobrecarga oral de glucosa o respuesta paradójica tras la administración de TRH13.
Los pacientes con acromegalia clínicamente activa se presentan generalmente como macroadenomas en las pruebas de imagen, igual que en los casos no familiares, mientras que aquellos clínicamente silentes con alteraciones bioquímicas suelen presentar pruebas de imagen negativas.
Se ha señalado que en numerosos casos de acromegalia clínicamente significativa, ésta no se hizo aparente hasta que fueron operados del síndrome de Cushing. El 72% de los pacientes presentan al mismo tiempo acromegalia y PPNAD.
Los hallazgos anatomopatológicos en pacientes con CNC y acromegalia muestran hiperplasia de células hipofisarias secretoras de GH y prolactina. Las células productoras de GH marcan positividad para prolactina y otras hormonas hipofisarias. La presencia de hiperplasia generalizada de células somatotropas en las autopsias realizadas a los pacientes con CNC son una explicación de la alta prevalencia de alteraciones bioquímicas de acromegalia. Análisis genéticos de estas lesiones muestran múltiples cambios que se acumulan de manera proporcional al grado de severidad del comportamiento clínico14.
TiroidesLa afectación de la glándula tiroidea abarca todo el espectro desde la patología nodular benigna hasta el carcinoma diferenciado de tiroides. Hasta un 75% de los pacientes presenta, por ecografía, enfermedad multinodular. El 25% presenta tumores tiroideos (siendo el más frecuente el adenoma folicular), y de ellos un 2,5% es de naturaleza maligna (carcinoma folicular o papilar). No existe riesgo aumentado de hipotiroidismo o hipertiroidismo15.
GónadasLa presencia de tumores testiculares aparece en un 33% de los pacientes afectados de CNC en el momento del diagnóstico. En su práctica totalidad se trata de tumores de células grandes calcificadas de Sertoli (large-cell calcifying Sertoli cell tumor, LCCSCT) y en casos excepcionales tumores de células de Leydig o restos de tumores adrenocorticales. La inmensa mayoría son tumores benignos no palpables con escaso potencial de malignidad. Son fácilmente diagnosticables por la presencia de microcalcificaciones bilaterales a través de ecografía. El LCCSCT puede ser hormonalmente activo y dar lugar a ginecomastia en varones prepuberales y peripuberales debido a un aumento de la expresión de la aromatasa periférica.
La presencia de quistes ováricos detectados por ecografía es un hallazgo frecuente. Generalmente son clínicamente insignificantes y de naturaleza benigna, aunque en casos excepcionales pueden desarrollar adenocarcinoma ovárico4,16.
TratamientoEl tratamiento en el CNC varía en función de las manifestaciones clínicas. Los tratamientos de elección son los siguientes:
- -
Mixoma cardíaco: cirugía cardíaca.
- -
Mixoma cutáneo: exéresis.
- -
Síndrome de Cushing: adrenalectomía bilateral.
- -
Adenoma hipofisario: cirugía transesfenoidal.
- -
Tumores tiroideos: cirugía, si se sospecha malignidad.
- -
LCCSCT: orquiectomía, especialmente en edad puberal y con ginecomastia.
- -
Schwanoma psamomatoso melanocítico: cirugía sobre lesión primaria y/o metastásica.
Los pacientes diagnosticados de CNC han de someterse a la realización de pruebas diagnósticas que permitan detectar la presencia de otras manifestaciones de la enfermedad. Es recomendable la realización de éstas con una periodicidad anual. Las pruebas recomendadas son las siguientes:
- -
Ecocardiograma anual.
- -
Determinación anual de cortisol libre urinario o test de Nugent.
- -
Determinación anual de IGF-1 plasmático.
- -
Ecografía de tiroides anual.
- -
Ecografía testicular anual en varones.
- -
Seguimiento clínico de adenoma ductal mama.
Para poder detectar de manera precoz la presencia de PPNAD o adenoma productor de GH es necesaria la realización de estudios más completos y específicos (test de Liddle de 6 días para PPNAD y sobrecarga oral de glucosa o test de TRH para la patología hipofisaria).
Genética del complejo de CarneyEl CNC es genéticamente heterogéneo. Se han identificado dos loci diferentes, uno en la región 17q22-24 (CNC1) y otro en la región 2p16 (CNC2)17,18. El gen causante del CNC1 fue identificado como subunidad reguladora (R1A) de la poteína kinasa A (PRKAR1A). En el CNC2 no ha sido hallado el gen causal. Mutaciones inactivantes heterocigóticas del gen PRKAR1A son las causantes de la enfermedad, encontrándose aproximadamente en un 70% de los pacientes. El 30% de los pacientes presenta una mutacion de novo y el resto presenta familiares afectados (70%).
El gen PRKAR1A comprende 11 exones y da lugar a una región genómica de aproximadamente 21 kb, con una región codificadora de 1.143 pares de bases (inicio en el exón 2)19–21.
La proteína expresada por el gen PRKAR1A es una de las subunidades reguladoras de la proteína kinasa A (PKA), una proteína kinasa dependiente de AMP cíclico (AMPc) clave en el proceso de señalización celular y relacionada en procesos de tumorogénesis endocrinológica, ya que está implicada en una amplia variedad de procesos celulares, incluyendo transcripción, metabolismo, progresión del ciclo celular y apoptosis.
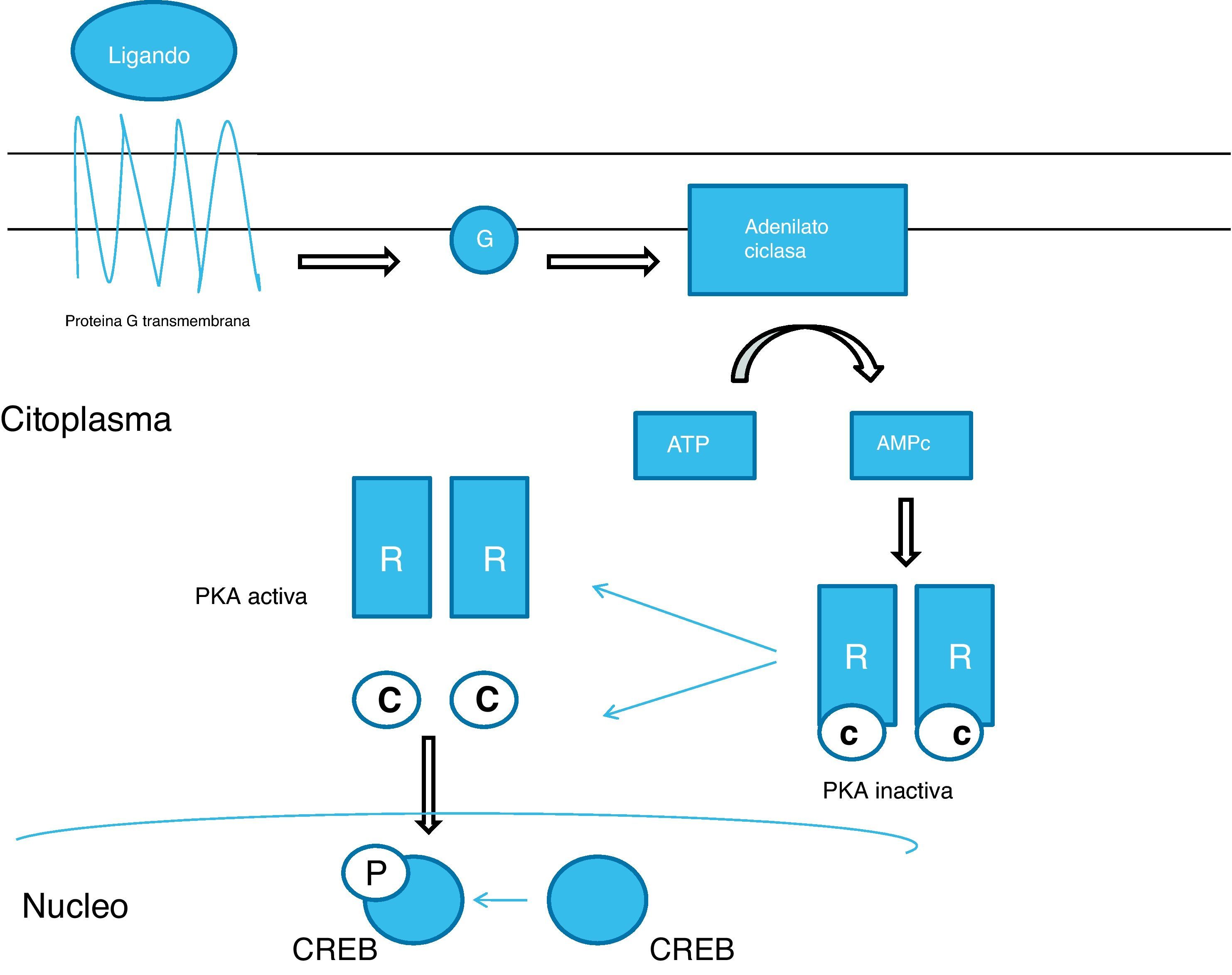
La PKA es un componente clave de la vía de señalización del AMPc. PKA es una holoenzima que comprende un tetrámero de dos unidades, con tres isoformas (C1, C2, C3), que son responsables de su actividad catalítica, y dos subunidades reguladoras que presenta cuatro isoformas (R1A, R1B, RIIA, RIIB) codificada por cuatro genes (PRKAR1A, PRKAR1B, PRKAR2A, PRKAR2B). Dependiendo de la combinación de estas subunidades, PKA se clasifica en tipo I (subunidad reguladora consistente en un dímero de R1A o R1B) y tipo II (subunidad reguladora consistente en un dímero de RIIA o RIIB)5. Por lo tanto, el gen PRKAR1A codifica la subunidad reguladora R1A que forma parte de la PKA. Ambos tipos de PKA se coexpresan en prácticamente todos los tejidos pero sus niveles de expresión varían entre tejidos. La PKA es inactiva en su forma tetramérica. La unión de AMPc a los dos sitios de unión situados en las subunidades reguladoras dan lugar a la disociación de las subunidades catalíticas de las subunidades reguladoras, dando lugar a la forma activa de la PKA. Esta forma activa es capaz de fosforilar residuos de serina o treonina a nivel de membrana, citoplasma o tras entrar en el núcleo la unidad catalítica, a nivel nuclear. La fosforilación a nivel nuclear del residuo ser-133 del CREB (c-AMP response element binding protein) produce la activación de factores de transcripción y consiguiente regulación de la expresión de genes downstream con el CRE (c-AMP response element)22,23 (fig. 1).
. Ésta interactúa con la adenilato ciclasa permitiendo la acumulación citoplasmática de AMPc. El AMPc entra en contacto con la proteína kinasa A (PKA) inactiva permitiendo la activación y liberación de las subunidades catalíticas (C). Estas subunidades catalíticas son capaces de fosforilar residuos de serina o treonina tanto en membrana, citoplasma o núcleo. Uno de estos targets de fosforilación por la PKA es el CREB (cAMP response element-binding) situado en el núcleo, que permite la activación de la transcripción.")
El ligando se une a la proteína G transmembrana activando la estimulación de la proteina G (G). Ésta interactúa con la adenilato ciclasa permitiendo la acumulación citoplasmática de AMPc. El AMPc entra en contacto con la proteína kinasa A (PKA) inactiva permitiendo la activación y liberación de las subunidades catalíticas (C). Estas subunidades catalíticas son capaces de fosforilar residuos de serina o treonina tanto en membrana, citoplasma o núcleo. Uno de estos targets de fosforilación por la PKA es el CREB (cAMP response element-binding) situado en el núcleo, que permite la activación de la transcripción.
En la serie internacional de Bertherat et al, un 73% de los pacientes con CNC presenta mutaciones inactivantes del gen PRKAR1A, y se detectan hasta 80 mutaciones diferentes. La mayor parte de las mutaciones son sustituciones de un par de bases o algunas inserciones o deleciones a nivel exónico. La gran mayoría son debidas a cambios de pauta de lectura (frameshift) debido a una inserción, deleción o algún cambio que afecta un par de bases, mientras que el resto fueron mutaciones de sentido erróneo (missense) o mutaciones sin sentido (nonsense) o afectando a un sitio de splicing. Estas mutaciones pueden afectar a cualquier región del gen aunque son más frecuentes en los exones 2, 3, 5, 7 y 84.
La mayoría de estas mutaciones (82%) da como resultado la aparición de un codón de terminación, que da lugar a RNAm anormal que provoca que la proteína que codifica este gen sea más corta de lo habitual (proteína truncada). Esta anomalía es detectada y reconocida por la nonsense mediated mRNA decay (NMD), que es activada por las células tisulares en el momento en que se detectan codificaciones de proteínas truncadas que impedirían el funcionamiento normal y da lugar a la degradación de este RNAm19. Esto significa que estas mutaciones no dan lugar a la expresión de una proteína anómala detectable (mutaciones no expresadas). Por lo tanto, a nivel molecular, la gran mayoría de las mutaciones presenta el mismo efecto: falta de una proteína mutada detectable y una reducción del 50% a nivel de expresión de la proteína R1A, dado que únicamente será capaz de expresarse el alelo no mutado. Este fenómeno conocido como haploinsuficiencia del gen PRKAR1A (incapacidad de que un solo gen normal mantenga el fenotipo normal de un individuo) se considera el responsable del CNC23,19,24.
A nivel bioquímico, la reducción de un 50% de R1A provoca un aumento compensatorio de los otros componentes del tetrámero de PKA, incluyendo las subunidades PKA tipo 1 (PRKAR1B) y tipo 2 (PRKAR2A y PRKAR2B) dependiendo del tejido, fase celular y otros factores, dando lugar a un aumento de la actividad PKA (provocada por la actividad de las subunidades catalíticas no controladas) y a anormalidades en el ciclo celular tanto en células humanas como de roedores24. En resumen, la inactivación del gen PRKAR1A está asociado con un aumento de la actividad PKA en determinados tejidos de origen endocrinológico donde puede estimular la proliferación celular dando lugar a la tumorogénesis endocrinológica característica del CNC25.
Recientemente se han descrito mutaciones en el gen PRKAR1A que escapan a la NMD y expresan proteínas mutadas a nivel celular, provocando una expresión clínica más agresiva debido a una mayor actividad PKA in vitro26,27. Con estos datos, surge la tentación de afirmar que el hecho de la actividad de la NMD ejerce un factor protector impidiendo una mayor expresividad clínica del CNC.
El hecho de que, a pesar del gran número de mutaciones descritas, la gran mayoría de ellas, por los mecanismos previamente descritos, presenta el mismo efecto final, explicaría la aparente falta de diferencias clínicas entre sujetos con diferentes mutaciones en el PRKAR1A y la aparente falta de presencia de correlación entre genotipo y fenotipo. Con el paso de los años se ha constatado, a pesar de la heterogeneidad propia del CNC, incluso en los pacientes que no han evitado la cascada de degradación, cierta relación entre genotipo y fenotipo. Las relaciones más destacadas serían la presencia de la mutación c.709_7del6 en los pacientes con PPNAD aislado, la mutación c.491_492del TG (mixomas cardíacos, lentiginosis y tumores tiroideos) y que la presencia de mixomas, tumores tiroideos, TGCGS y schwanoma son más frecuentes en aquellos pacientes portadores de mutaciones en el gen PRKAR1A que en aquellos no portadores. Como curiosidad, la mutación más frecuente descrita es c.491_492 del TG, observada en varias familias no relacionadas4.
La determinación de mutaciones inactivantes de la subunidad reguladora tipo 1A (R1A) del gen de la PKA (PRKAR1A) en la mayoría de los pacientes con CNC ha permitido una estimación de su penetrancia, la validación de los criterios diagnósticos de la enfermedad y la constatación de que no existe una clara relación fenotipo-genotipo.
Consejo genéticoEl estudio genético para determinación de mutaciones inactivantes debería ser propuesto a todos los casos índice. Si se detecta una mutación del gen PRKAR1A, debería proponerse estudio genético a los familiares de primer grado. Esto permitiría un diagnóstico precoz y realización de pruebas para buscar otras manifestaciones del CNC en pacientes portadores y evitar pruebas innecesarias a los familiares no portadores con la consiguiente reducción del gasto sanitario y de la ansiedad en estos sujetos28–29.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
