


Cystic fibrosis is the most common fatal inherited autosomal recessive disease in Caucasians, affecting approximately one out of every 2000 births. Survival of patients with cystic fibrosis has significantly improved due to advances in respiratory and nutritional care, and their current average life expectancy is 30–40 years. Development of cystic fibrosis-related diabetes is a comorbidity that increases with age and may reach a prevalence up to 50% in adults. Its development is associated to impaired lung function and nutritional status, and early diagnosis and treatment are therefore essential to improve quality of life and performance status. Insulin therapy for diabetes and other early carbohydrate metabolism disorders may improve lung function and nutritional status of patients with cystic fibrosis.
La fibrosis quística es la enfermedad genética letal con herencia autosómica recesiva más frecuente en la raza caucásica, afectando aproximadamente a uno por cada 2.000 nacidos vivos. La supervivencia de los pacientes con fibrosis quística ha mejorado gracias a los avances en los cuidados respiratorios y nutricionales, alcanzando un promedio de esperanza de vida comprendido entre los 30 y 40 años. La aparición de diabetes en la fibrosis quística es una comorbilidad que aumenta con la edad, con una prevalencia de hasta el 50% en los adultos. Su desarrollo se asocia a un empeoramiento de la función pulmonar y el estado nutricional, por lo que su diagnóstico precoz es esencial para mejorar la calidad de vida y clase funcional. El tratamiento con insulina de la diabetes y otras alteraciones precoces del metabolismo hidrocarbonado podría conllevar a una mejora de la función pulmonar y del estado nutricional de los pacientes con fibrosis quística.
The incidence of cystic fibrosis (CF) in Spain ranges from 1:2810 to 1:5532 live births depending on the study. Such variation is probably due to the different prevalence rates of the main mutation, F508¿, consisting of the disappearance of a phenylalanine at position 508 of the CFTR (cystic fibrosis transmembrane regulator) protein. The frequency of this mutation as the cause of the disease in Spain ranges from 50% in Mediterranean populations and to 80% in Asturias.1–3 Because of the longer survival of patients with CF, cystic-fibrosis related diabetes (CFRD) is currently one of the most common comorbidities in subjects who reach adulthood. The first cases reported of CFRD and glucose intolerance (GI) in CF date back to 1955.4
CFRD is typically diagnosed in late adolescence, with a mean age at diagnosis ranging from 18 to 25 years. It is difficult to determine its prevalence and, because of the diagnostic and monitoring criteria used, it is often underestimated. The overall prevalence rates estimated in Europe and the United States are 12.4% and 16.9%, respectively.5,6 CFRD may occur at any age, but prevalence most commonly increases with age from 9% at 5–9 years, 26% at 10–20 years, to up to around 50% at 30 years.4,7 From 15 to 30 years of age, the proportion of patients with normal glucose tolerance decreases almost linearly, although it should be noted that this is a difficult estimation because metabolic status may change from one year to the other in patients with CF.8–11
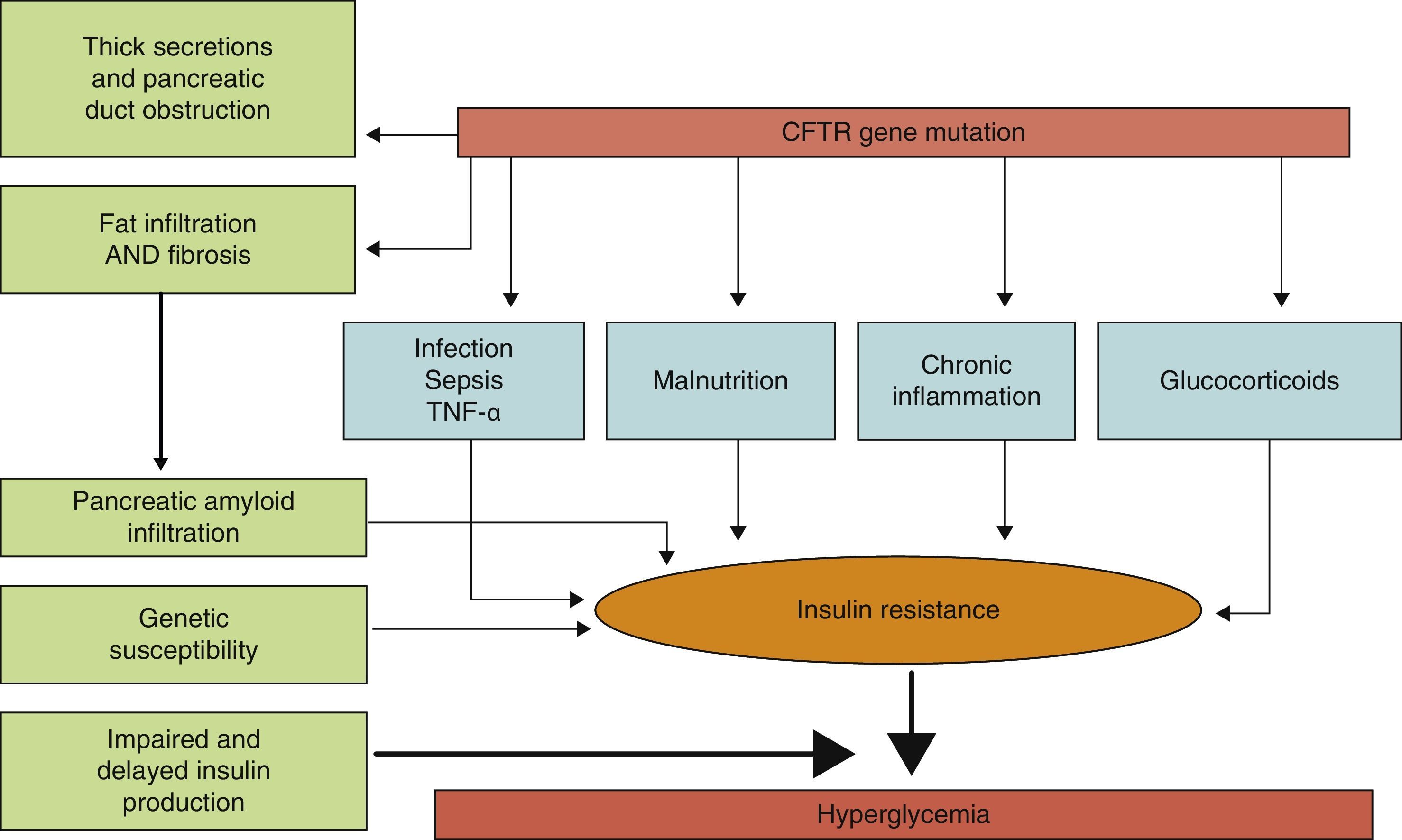
PathophysiologyLittle is known about the factors which predict for the development of diabetes in CF, and it is not clear which characteristics differentiate subjects with CF who will or will not develop diabetes. The predisposing factors identified include (Fig. 1) female sex, age, type of genetic mutation (a greater risk in people homozygous for the F508¿ mutation), exocrine pancreatic insufficiency, the degree of pulmonary function impairment, the use of corticosteroids, and the presence of other concurrent complications of CF such as liver disease and lung transplant.8,12

Physiopathogenic mechanisms of carbohydrate metabolism impairment in CF.
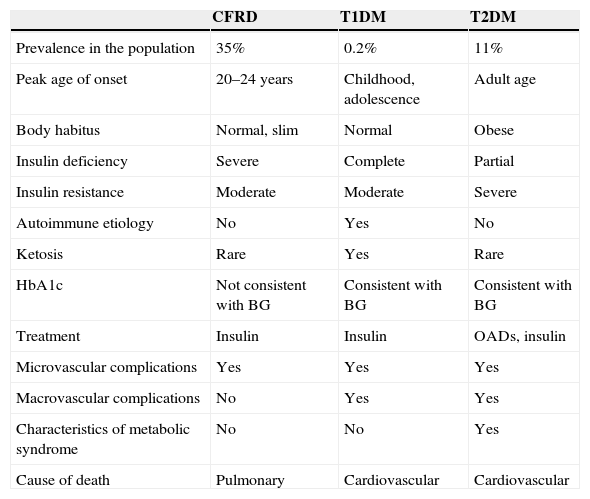
The exact mechanism by which diabetes occurs in CF has still to be elucidated. It is known that the main physiopathogenic factor is insulin deficiency associated with ¿-cell dysfunction, but other factors such as insulin resistance, impaired function of other pancreatic hormones, and impairment in the enteroinsular axis and insulin clearance also have an influence.10,13 Although CFRD has similarities to type 1 and 2 diabetes mellitus (Table 1), it should be considered as a different condition, with specific long-term goals.11
Main characteristics of CFRD as compared to type 1 and type 2 DM.
| CFRD | T1DM | T2DM | |
|---|---|---|---|
| Prevalence in the population | 35% | 0.2% | 11% |
| Peak age of onset | 20–24 years | Childhood, adolescence | Adult age |
| Body habitus | Normal, slim | Normal | Obese |
| Insulin deficiency | Severe | Complete | Partial |
| Insulin resistance | Moderate | Moderate | Severe |
| Autoimmune etiology | No | Yes | No |
| Ketosis | Rare | Yes | Rare |
| HbA1c | Not consistent with BG | Consistent with BG | Consistent with BG |
| Treatment | Insulin | Insulin | OADs, insulin |
| Microvascular complications | Yes | Yes | Yes |
| Macrovascular complications | No | Yes | Yes |
| Characteristics of metabolic syndrome | No | No | Yes |
| Cause of death | Pulmonary | Cardiovascular | Cardiovascular |
BG: blood glucose (plasma glucose); T1DM: type 1 diabetes mellitus; T2DM: type 2 diabetes mellitus; CFRD: cystic fibrosis-related diabetes.
Source: Laguna et al.11
The first phase of insulin secretion in response to an oral and intravenous glucose overload (and to other insulin secretion stimuli) is impaired in adults with CF.9,10,14,15 The insulin peak is significantly delayed by up to 90–120min in patients with CFRD, as compared to 30–60min in healthy subjects.10 This loss of the first phase of insulin secretion occurs even in subjects with CF and normal glucose tolerance (NGT).9,14,15 The absolute amount of insulin secreted over time is also decreased in patients with CF.6,9 This overall decrease in insulin secretion has been associated with the presence of exocrine pancreatic insufficiency. Patients with exocrine pancreatic insufficiency have a 41% decrease in peak plasma insulin in response to glucose intake. Glucagon and pancreatic polypeptide secretion is also lower, while somatostatin levels may be higher. Glucagon response to hypoglycemia is globally decreased in patients with CF, both those with CFRD and those with a normal carbohydrate metabolism. It has been suggested that this insulinopenia associated with changes in glucagon and other pancreatic hormones slows the development of CFRD and makes episodes of ketoacidosis uncommon in this type of diabetes.10 To sum up, peak insulin secretion, the first phase of insulin secretion, and the total amount of insulin secreted in response to a glucose overload are decreased in CFRD.
An additional factor that may be involved in CFRD occurrence is insulin resistance (IR). The pathogenic role of IR is controversial, but it appears to be widely accepted that patients with CFRD have different grades of IR, and that some clinical conditions such as infections, nutritional status, the use of corticosteroids, and the clinical state of pulmonary disease may lead to a significant exacerbation of hyperglycemia. As regards the mechanisms of IR in CFRD, changes in the GLUT-4 transporter have been reported.15,16 Patients with CF have low-grade chronic inflammation, and TNF¿ elevation has therefore also been postulated as a mechanism promoting IR. There are studies showing that TNF¿ levels are increased in plasma and bronchoalveolar lavage samples from patients with CF, and that they correlate to clinical status scoring systems, with a trend to higher levels in subjects with CFRD and GI.16 More recently, levels of gamma-glutamyl transpeptidase, another marker of IR, have been seen to be apparently related to an increased risk of CFRD regardless of age, sex, and body fat.12 Similarly, some studies in patients with CF have found high levels of free fatty acids, closely related to IR and associated with the development of CFRD.10
DiagnosisDiabetes in CF is currently diagnosed using the criteria of the North American Cystic Fibrosis Foundation and the American Diabetes Association (ADA) based on glycosylated hemoglobin (HbA1c), fasting plasma blood glucose, or the 2-hour oral glucose tolerance test (OGTT).15–19
HbA1c (%) should not be used as a screening test for CFRD because it may be normal in the early stages and in cases of intermittent hyperglycemia. Moreover, because of the chronic inflammation state, erythrocyte half-life is decreased in CF, and HbA1c values may therefore be lower than expected.9,14,15,17 Holl et al.18 conducted a study showing that mean plasma glucose levels at 120min were not different in cases of CF with elevated HbA1c as compared to those with normal values, and concluded that HbA1c should not replace the OGTT as a screening test because of its low sensitivity.
The OGTT is currently the most sensitive screening test for detecting CFRD, and the one recommended by the International Society for Pediatric and Adolescent Diabetes and the ADA. This is because if plasma glucose levels are only tested under fasting conditions, more than two-thirds of patients with CFRD may go undiagnosed.
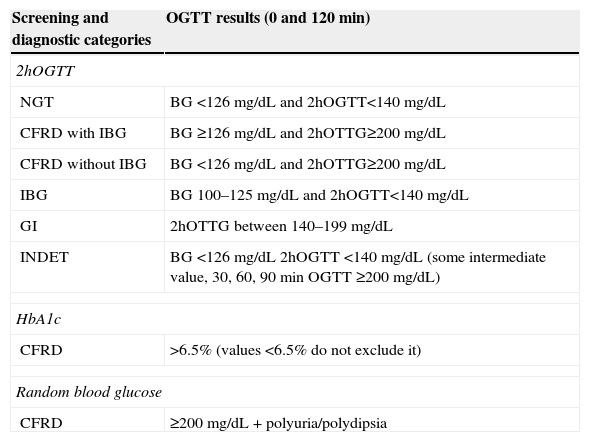
Based on the OGTT results, patients may be classified into several categories (Table 2) depending on blood glucose levels at different times during the test: NGT, CFRD with impaired basal glucose (IBG), CFRD with no IBG, impaired fasting blood glucose (FBG), GI and indeterminate glucose tolerance (INDET).4,9,17,19
Diagnostic categories of carbohydrate metabolism changes according to the results of the oral glucose tolerance test (OGTT). North American CF Consensus Conference 2010.17,19
| Screening and diagnostic categories | OGTT results (0 and 120min) |
|---|---|
| 2hOGTT | |
| NGT | BG <126mg/dL and 2hOGTT<140mg/dL |
| CFRD with IBG | BG ≥126mg/dL and 2hOTTG≥200mg/dL |
| CFRD without IBG | BG <126mg/dL and 2hOTTG≥200mg/dL |
| IBG | BG 100–125mg/dL and 2hOGTT<140mg/dL |
| GI | 2hOTTG between 140–199mg/dL |
| INDET | BG <126mg/dL 2hOGTT <140mg/dL (some intermediate value, 30, 60, 90min OGTT ≥200mg/dL) |
| HbA1c | |
| CFRD | >6.5% (values <6.5% do not exclude it) |
| Random blood glucose | |
| CFRD | ≥200mg/dL+polyuria/polydipsia |
NGT: normal glucose tolerance; CFRD with IBG: cystic fibrosis-related diabetes with impaired blood glucose; CFRD without IBG: cystic fibrosis-related diabetes without impaired blood glucose; IBG: impaired blood glucose; HbA1c: glycosylated hemoglobin; INDET: indeterminate glucose tolerance; GI: glucose intolerance; 2hOGTT: oral glucose tolerance test, with blood glucose measurements at 0 and 120min.
The natural course of diabetes in CF is first characterized by the occurrence of intermittent postprandial hyperglycemia, followed by GI, CFRD with no IBG and, finally, CFRD with IBG. Thus, approximately 15% of the adults with CFRD have IBG, and 25% have no IBG.14,15 It should be noted that IBG occurs in a lower proportion of patients as compared to other types of diabetes. Cases of fasting hypoglycemia have even been reported.20,21 That is, there is a lower overall prevalence of IBG.22,23
An annual OGTT during a phase of clinical stability is usually recommended in patients over 10 years of age,19 although screening before that age has recently been suggested. Odet et al.24 studied 94 children with CF aged 6–9 years and found an impaired carbohydrate metabolism in 41% of them; up to 42% of these children developed diabetes. According to these results, children younger than 10 years with CF already have an increased risk of developing diabetes. The OGTT should also be performed in pregnant women, before transplantation, and in patients with impaired lung function, delayed growth or pubertal development, unexplained weight loss, and classical symptoms of diabetes or hypoglycemia. Fasting and postprandial capillary blood glucose levels should be monitored in the event of hospitalization for severe disease, artificial nutrition, the use of corticosteroids, and before and after major surgery. Patients with CFRD but no IBG, or with GI, who are at risk of progressing to CFRD with IBG, should be studied annually.14,15,19 Symptoms of diabetes are not a sensitive indicator, because their occurrence is insidious and very late.
As regards the OGTT, the hyperglycemic peak at 120min in most patients with CF may be absent, and sampling every 30min is therefore considered necessary. Subjects with CF but no CFRD have been seen to experience hyperglycemia at 30, 60, and 90min, but blood glucose levels at 0 and 120min are similar to those of healthy controls. Therefore, blood glucose levels at 60 and 90min may be more sensitive for detecting glucose intolerance in these patients.21,22 Normal random blood glucose levels do not rule out diagnosis, because they depend on the time of day, the last meal, gastric emptying, etc. By contrast, persistently high levels (≥200mg/dL) allow for diagnosis, and the ADA has therefore incorporated random blood glucose levels as a diagnostic tool.25 In this regard, one option is to obtain a glycemic profile using multiple capillary blood glucose measurements over a long period and varying the times at which the samples is taken. Recent studies26 have found an association between worse lung function and the presence of hyperglycemia at intermediate points in the OGTT, specifically with blood glucose levels at 60min higher than 140mg/dL. By contrast, this same study did not find a negative correlation between forced expiratory volume in one second (FEV1%) and blood glucose levels at 120min, and suggested that a diagnosis of carbohydrate tolerance status should not depend on blood glucose levels at the conventional times of 0 and 120min. Lee et al.27 analyzed the diagnostic value of the 1h-Glucose Challenge Test (GCT). This is a simpler test with an oral glucose overload of 50g and plasma glucose measurements before and one hour after glucose intake. They considered plasma glucose levels higher than 140mg/dL one hour after glucose overload to be positive. Thirty-one patients with CF and no prior history of CFRD were given a GCT, and an OGTT one week later. Both tests detected hyperglycemia in 30% of the subjects (all patients with hyperglycemia in the OGTT were also detected in the GCT), while hyperglycemia was identified the GCT in only 35%. These results led the authors to suggest that the OGTT (considered to be the gold standard test) may underestimate the presence of short hyperglycemic events (blood glucose >140mg/dL) in a significant proportion of cases, but additional studies are needed in this population to assess the diagnostic value of the GCT. Recently, Schmid et al.28 analyzed the prevalence of a glucose peak higher than 200mg/dL at some intermediate point in the curve. They found that, when the overall series was considered, the INDET thus defined affected 10% of the patients, and 22.2% of those initially classified as having NGT. These authors therefore support the concept of the importance of considering intermediate changes in the OGTT when studying patients with CF.17,29 New approaches are now available to better characterize changes in carbohydrate metabolism in patients with NGT based on increased plasma insulin and C-peptide levels during the OGTT. Patients with lower basal levels and lower increases in such parameters during the OGTT have poorer nutritional status and more impaired lung function.29
Mortality and lung functionAlthough CFRD shares some characteristics of type 1 and 2 DM, it also shows marked differences that make its diagnosis, management, and treatment specific. The early diagnosis of changes in carbohydrate metabolism is important in patients with CF because diabetes development is associated with a 31–55% increase in the risk of death, irrespective of other complications of CF.30 Insulin deficiency promotes a negative protein balance, contributing to the morbidity and mortality of these subjects with increased catabolism and weight loss.9 Weight loss may occur even with early changes in carbohydrate metabolism.23
Other factors associated with increased mortality include age, female sex, impaired lung function, malnutrition, lung infection by the Burkholderia cepacia complex, the diagnosis of allergic bronchopulmonary aspergillosis, liver disease, and a prior organ transplant.30
Respiratory disease accounts for most morbidity and mortality and is, together with malabsorption, the most common presentation of the disease. Patients with CFRD have a greater decline in lung function as compared to normoglycemic patients, which is directly related to the degree of insulin deficiency and to the severity of carbohydrate metabolism impairment. In other words, the degree of glucose intolerance is directly correlated to lower FEV1% and forced vital capacity (FVC%) vaues.15 CFRD is even associated with a poorer score in the scales used30 to define the clinical and radiographic stage of lung disease, such as the Shwachman and Kulczycki31 and Brasfield32–34 scales.
The impairment in lung function parameters is often seen before the diagnosis of CFRD. In children, such impairment has been found up to one year before. Other authors report that this decline in lung function may be seen up to three or four years before CFRD occurs.35 This time interval may be explained by the changes in protein catabolism associated with insulin deficiency and by the resultant decrease in muscle mass, with a consequent decrease in inspiratory function of the diaphragm.15,17 Histological studies of the lungs of diabetic patients with no CF have shown the presence of fibrosis and the thickening of basal lamina. Other factors mentioned in the literature as being responsible for lung disease in diabetic patients with no CF include non-enzymatic abnormal glycosylation of bronchial tree proteins, the presence of underlying pulmonary microangiopathy, and increased bronchial reactivity as a consequence of autonomic and phrenic neuropathy.36,37
Severe lung disease (defined as FEV1 <40%) may be associated with poor blood glucose control in subjects with CFRD. On the other hand, diabetes is also associated with an increased frequency of exacerbations of pulmonary disease and an increased prevalence of pathogens in sputum.8,15 Colonization by Pseudomonas aeruginosa is also associated with a poorer clinical outcome and lung function, more hospital admissions and lower weight, and is a significant predictor of clinical impairment and morbidity and mortality.38–40 As a consequence, patients with CFRD have decreased survival and poorer quality of life.
Microvascular and macrovascular complications in cystic fibrosis-related diabetesMicrovascular complications have been reported in studies of small series of patients with CFRD. Patients with CFRD without IBG did not experience microvascular complications in any of these studies during follow-up periods of up to 14 years. Although it has been suggested that patients with CFRD without IBG do not require treatment because they are symptom-free and do not appear to run a risk of microvascular complications, the nutritional consequences of insulin deficiency may impair their quality of life.23 Among patients with CFRD and IBG followed up for longer than 10 years, microalbuminuria occurred in 14% of cases, retinopathy in 16%, neuropathy in 55%, and gastropathy in 50%. The ADA recommends annual monitoring for microvascular complications from 5 years after the diagnosis of CFRD.19 According to a study by Schwarzenberg et al., the most common complication is neuropathy, with a similar prevalence to that found in type 1 and 2 DM. The most common finding is a decreased amplitude of sural nerve action potential, together with impaired cardiorespiratory reflexes.5 In another study comparing the frequency of microvascular complications in a group of patients with CFRD to another group with type 1 DM, microalbuminuria was more prevalent in the CFRD group (with normal kidney function), while retinopathy was more common in patients with type 1 DM. No differences were found in other microvascular complications such as peripheral neuropathy.41,42
Few reports or series of CF patients with macrovascular complications are available, probably because of the short overall life expectancy of this population and the low rate of dyslipidemia, hypertension, and obesity.
TreatmentTreatment has a dual objective: to improve the nutritional status of patients with the anabolic effect of insulin–with a resultant improvement in lung function and to decrease the incidence of late complications of diabetes, particularly microvascular complications.
Insulin therapy is currently recommended for CFRD with IBG.19 The use of oral antidiabetic drugs is not recommended.14,19 There is little evidence of the superiority of specific insulin regimens in CFRD. Insulin therapy is also indicated in the presence of cardinal diabetes symptoms (polydipsia, polyuria, weight loss), unexplained weight loss despite adequate nutritional management, delayed growth and puberty, or an unexplained decline in lung function.14 However, the results of a clinical trial where treatment with preprandial rapid-acting insulin reversed the trend to gradual weight loss in patients with CFRD without IBG, increasing body fat and mass, have recently been published.43 The results of this study would suggest the benefit of insulin therapy in patients with CFRD (with and without fasting hyperglycemia). The benefits of insulin therapy include increased weight, improved lung function (FEV1%), and decreased pulmonary exacerbations. Treatment should be individualized and adjusted to carbohydrate intake, which should not be restricted in order to improve blood glucose control. It is, however, not clear what should be the approach in patients with CF and GI or INDET should be, or whether the early treatment of carbohydrate changes with insulin would be able to decrease the gradual impairment in lung function and nutritional status. Some recently reported studies, of a small number of patients, have shown that early insulin treatment in these situations is also beneficial.44–46 Bizzarri et al.47 studied the results of a daily dose of insulin glargine in patients with CF and GI. The authors showed that this treatment resulted in a significant increase in the body mass index and FEV1% during the follow-up period of the study (1–1, 8 years), and no episode of hyperglycemia occurred. They postulated that early treatment with insulin is well tolerated and could decrease the decline in lung function and nutritional status.47 Other authors, such as Dobson et al., studied the effect of insulin therapy in patients with long-standing CF with lung function and weight impairment with no clear cause and with normal OGTT and HbA1c, but with postprandial hyperglycemic peaks higher than 180mg/dL, detected in capillary blood glucose monitoring at home. Insulin therapy increased weight in all patients and improved lung function, with no symptoms or blood glucose values suggestive of hypoglycemia.46 Mozillo et al.22 recently reported the effects of treatment with insulin glargine for one year in a group of CF patients with different carbohydrate metabolism changes. After one year of insulin therapy, an overall significant improvement in FEV1% and a decrease in the number of pulmonary exacerbations were seen.22
Thus, the above discussed studies suggested that treatment with insulin of different early changes in carbohydrate metabolism should be considered as a therapeutic option for improving lung function and the quality of life of this group of patients. However, despite these initial positive data regarding insulin therapy in early glucose metabolism changes in CF patients, additional controlled, randomized studies with longer observation periods are needed to assess the pulmonary and nutritional effects of insulin in patients with early carbohydrate metabolism changes such as GI and INDET.
ConclusionsBecause of the improved diagnostic procedures and treatments available, patients with CF are now expected to live until the fourth or fifth decades of life, and CFRD is therefore becoming increasingly prevalent.
The onset of diabetes is associated with increased morbidity and mortality, and with impaired lung function, nutritional status, and a functional clinical state use. CFRD should be considered a specific condition different from type 1 or 2 DM whose early diagnosis is essential in order to start measures to control its specific harmful effects on the underlying disease.
Today, diagnostic criteria for carbohydrate changes in CF are based on the OGTT timepoints of 0 and 120min, but increasing clinical significance is recently being attributed to hyperglycemia in the intermediate timepoints of the test (30, 60, and 90min), which various studies relate to the functional prognosis of CF and which should therefore be taken into account in the overall assessment of patients with carbohydrate changes.
Early insulin therapy for CFRD, and probably other early glucose metabolism changes, should lead to improvements in the pulmonary and nutritional status and the quality of life of these patients.
Conflicts of interestThe authors state that they have no conflicts of interest.
Please cite this article as: Cano Megías M, González Albarrán O. Diabetes en la fibrosis quística: una entidad diferente. Endocrinol Nutr. 2015;62:38–44.
