



El carcinoma medular de tiroides es un tumor de baja prevalencia cuyo pronóstico es peor que el del cáncer diferenciado de tiroides debido su mayor agresividad. El objetivo de este trabajo es describir las características demográficas, clínicas y genéticas de los pacientes atendidos en el área sanitaria de la Comunidad de Castilla-La Mancha durante 16 años.
Pacientes y métodosLos datos se recogieron mediante revisión de historias clínicas.
ResultadosSe revisaron las historias clínicas de 58 pacientes con una edad media al diagnóstico de 51 años (intervalo de 6 a 82 años) y un 63,8% de mujeres. La prevalencia fue de 2,84 casos por 100.000 habitantes, con una gran variabilidad entre áreas (de 0 a 5,4 casos por 100.000 habitantes). Los casos familiares representaron el 34,5% del total, siendo la mutación más frecuente la C634Y. El motivo más frecuente de diagnóstico fue la palpación de un bultoma cervical (70,6%); se solicitó ecografía al diagnóstico en 56 de 58 casos, y la calcitonina en 8 de 58 casos. La multicentricidad del tumor fue descrita en el 59 y 50% de los casos de síndrome de neoplasia endocrina múltiple tipo 2A y 2B, respectivamente, y en ningún caso esporádico. El 52% de los pacientes presentaba un estadio avanzado al diagnóstico (iii o iv). La mediana de seguimiento fue de 36 meses (rango intercuartílico 14-210), con la pérdida de 11 pacientes durante el seguimiento.
ConclusionesEl diagnóstico de carcinoma medular de tiroides en Castilla-La Mancha se basa en la ecografía cervical, pero no en la calcitonina. Existe una alta prevalencia de este carcinoma, tanto familiar como esporádico, y una importante variabilidad en el tipo de mutación del protooncogén rearranged during transfection comparadas con las del resto de la población española.
Medullary thyroid cancer is a rare tumor that is more aggressive and has a worse prognosis than differentiated thyroid cancer. The purpose of this study was to report the demographic, clinical, and genetic characteristics of patients seen in the health care system of the community of Castilla-La Mancha over a 16-year period.
Patients and methodsData were collected through a review of patients’ medical records.
ResultsThe medical records of 58 patients (mean age at diagnosis, 51 years; range, 6-82 years; 63.8% women) were reviewed. Prevalence rate was 2.84 cases per 100,000 inhabitants, with a high variability between areas (range, 0-5.4 cases per 100,000 inhabitants). Familial cases accounted for 34.5% of all medullary thyroid cancers, and the most common mutation was C634Y. The condition was most commonly diagnosed following palpation of a cervical lump (70.6%). At diagnosis, 56 of 58 patients underwent ultrasound and 8 of 58 patients were tested for serum calcitonin. Tumor multicentricity was reported in 59 and 50% of patients with multiple endocrine neoplasia syndrome type 2A and 2B, respectively, and in no sporadic cases. Fifty-two percent of patients had an advanced stage (iii or iv) at diagnosis. Median follow-up was 36 months (interquartile range, 14-210); 11 patients were lost to follow-up.
ConclusionsIn Castilla-La Mancha, medullary thyroid cancer is diagnosed by cervical ultrasound, rather than calcitonin assay. There is a high prevalence of both familial and sporadic medullary thyroid cancer, and a significant variability in the type of proto-oncogen rearranged during transfection mutation as compared to the rest of the Spanish population.
El carcinoma medular de tiroides (CMT) es un tumor raro que representa entre el 5 y el 10% de todos los cánceres tiroideos1. Se origina a partir de las células parafoliculares o células C de la glándula tiroidea, y produce una hipersecreción de calcitonina como señal bioquímica precoz. Puede ser esporádico o formar parte del síndrome de neoplasia endocrina múltiple tipo 2 (MEN-2), que tiene un patrón de herencia autosómico dominante. El CMT invade fácilmente los ganglios linfáticos intraglandulares y se expande por el resto de la glándula tiroidea, además de hacerlo hacia los ganglios linfáticos pericapsulares y regionales. El diagnóstico en estadios iniciales se asocia a un mejor pronóstico2. Debido a la baja prevalencia de esta entidad, el análisis de todos los casos diagnosticados es esencial para poder ampliar su conocimiento. En este sentido, el presente estudio aporta por primera vez información sobre la situación del CMT en Castilla-La Mancha.
El objetivo de este trabajo es describir las características demográficas y clínicas de una serie de pacientes con CMT atendidos en las 8 áreas sanitarias de la Comunidad de Castilla-La Mancha, así como analizar las diferencias entre los casos esporádicos y los familiares.
Pacientes y métodosSe trata de un estudio observacional, retrospectivo y multicéntrico, promovido por la Sociedad Castellano Manchega de Endocrinología, Nutrición y Diabetes, que incluye a todos los pacientes con diagnóstico anatomopatológico de CMT entre los años 1995 y 2010, ambos inclusive. No fueron objeto de este estudio los pacientes con diagnóstico anatomopatológico de hiperplasia de células C. La selección de casos se realizó a partir de las bases de datos de los servicios de Anatomía Patológica de las 8 áreas sanitarias de Castilla-La Mancha (Albacete, Ciudad Real, Cuenca, Guadalajara, Mancha-Centro, Puertollano, Talavera de la Reina y Toledo), detectándose todos los casos cuyo diagnóstico citológico o histológico incluyera las palabras «carcinoma medular de tiroides», a excepción del Hospital de Manzanares, que no participó en el estudio. La recogida de datos se realizó entre enero y septiembre de 2011 mediante la revisión de la historia clínica de los pacientes anteriormente seleccionados.
El estudio fue aprobado por el Comité de Ética y por la Comisión de Investigación Clínica del Complejo Hospitalario de Toledo como centro coordinador del estudio.
Variables analizadasPara cada paciente se recogieron las siguientes variables:
- 1.
Variables sociodemográficas: edad, sexo y área sanitaria de referencia.
- 2.
Variables clínicas: edad al diagnóstico, motivo del diagnóstico, duración del seguimiento expresada en meses, técnicas de imagen solicitadas para el diagnóstico, determinación de calcitonina al diagnóstico (sí/no), multicentricidad en la pieza quirúrgica (sí/no), presencia de metástasis y su localización, estadio al diagnóstico basado en la clasificación TNM y el tratamiento realizado (quirúrgico y/o médico).
- 3.
Estudio genético: clasificación fenotipo-genotipo y tipo de mutación en el gen REarranged during Transfection (RET), si está presente. Los casos referidos como esporádicos fueron casos sin mutación en el gen RET.
La prevalencia se estimó basándose en los datos poblacionales del estudio DIACAM1, que fue objeto de una publicación anterior3. La presencia de metástasis a distancia se documentó mediante su notificación en la historia clínica y las pruebas de imagen.
No fueron objeto de este estudio las características ecográficas, citológicas ni anatomopatológicas de los tumores, a excepción de la multicentricidad en la pieza quirúrgica.
Análisis estadísticoLas variables cuantitativas se expresan como media y desviación típica si se ajustan a la normalidad, o mediante la mediana y el rango o intervalo intercuartílico (RI) en caso contrario. Las variables cualitativas se expresan como porcentaje. Se utilizaron pruebas paramétricas como la t de Student para la comparación de medias y la χ2 para la comparación de variables cualitativas. El umbral de significación que se adoptó para todas las pruebas fue p<0,05. El análisis de los datos se realizó con el paquete estadístico SPSS® v.11.0.
ResultadosSe revisaron las historias clínicas de 58 pacientes diagnosticados de CMT entre 1995 y 2010. En 2 casos no se realizó tratamiento quirúrgico debido a la presencia de enfermedad localmente avanzada, por lo que el diagnóstico en esos 2 casos fue mediante citología con punción-aspiración con aguja fina. En el resto de los casos el diagnóstico fue histológico mediante análisis de las piezas de tiroidectomía.
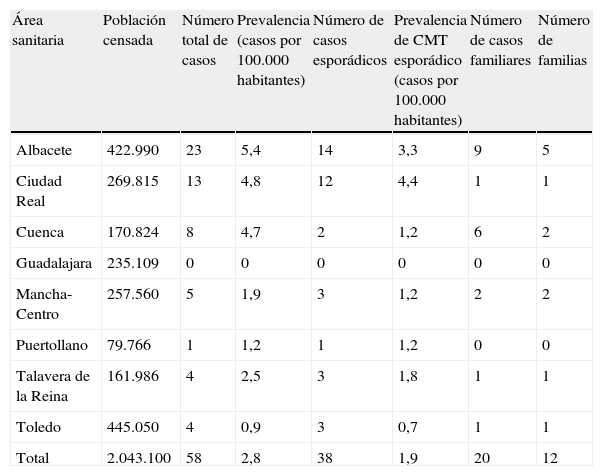
PrevalenciaLa prevalencia media de CMT en nuestra cohorte fue de 2,84 casos por 100.000 habitantes, con una gran variabilidad entre las distintas áreas sanitarias tanto para casos esporádicos (4,4 y 3,3 casos/100.000 habitantes en las áreas de Ciudad Real y Albacete frente a 0 y 0,7 casos/100.000 habitantes en las áreas de Guadalajara y Toledo) como familiares (9 casos pertenecientes a 5 familias en el área de Albacete frente a ningún caso familiar en las áreas de Guadalajara o Puertollano) (tabla 1).
Prevalencia del carcinoma medular tiroideo
| Área sanitaria | Población censada | Número total de casos | Prevalencia (casos por 100.000 habitantes) | Número de casos esporádicos | Prevalencia de CMT esporádico (casos por 100.000 habitantes) | Número de casos familiares | Número de familias |
| Albacete | 422.990 | 23 | 5,4 | 14 | 3,3 | 9 | 5 |
| Ciudad Real | 269.815 | 13 | 4,8 | 12 | 4,4 | 1 | 1 |
| Cuenca | 170.824 | 8 | 4,7 | 2 | 1,2 | 6 | 2 |
| Guadalajara | 235.109 | 0 | 0 | 0 | 0 | 0 | 0 |
| Mancha-Centro | 257.560 | 5 | 1,9 | 3 | 1,2 | 2 | 2 |
| Puertollano | 79.766 | 1 | 1,2 | 1 | 1,2 | 0 | 0 |
| Talavera de la Reina | 161.986 | 4 | 2,5 | 3 | 1,8 | 1 | 1 |
| Toledo | 445.050 | 4 | 0,9 | 3 | 0,7 | 1 | 1 |
| Total | 2.043.100 | 58 | 2,8 | 38 | 1,9 | 20 | 12 |
CMT: carcinoma medular de tiroides.
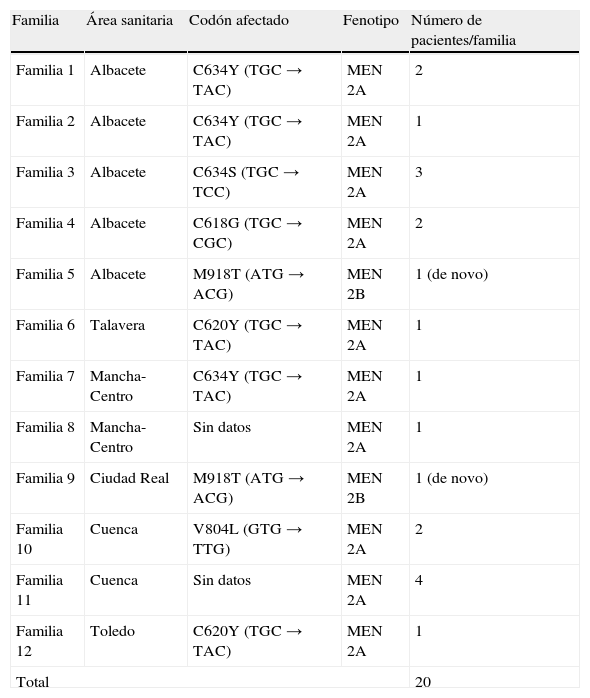
El 63,8% eran mujeres, con una relación hombre/mujer de 1/1,76. La edad media al diagnóstico fue de 50,82 años (intervalo de 6 a 82 años), sin diferencia estadística entre sexos. La proporción de casos esporádicos frente a los familiares fue de 38/20, representando los casos familiares el 34,5% del total estudiado (18 pacientes MEN 2A y 2 pacientes MEN 2B). Las mutaciones más frecuentes fueron la C634Y (3 de 9 familias) y la C620Y (2 de 9 familias) (tabla 2).
Genotipo-fenotipo en los casos de carcinoma medular tiroideo familiar
| Familia | Área sanitaria | Codón afectado | Fenotipo | Número de pacientes/familia |
| Familia 1 | Albacete | C634Y (TGC→TAC) | MEN 2A | 2 |
| Familia 2 | Albacete | C634Y (TGC→TAC) | MEN 2A | 1 |
| Familia 3 | Albacete | C634S (TGC→TCC) | MEN 2A | 3 |
| Familia 4 | Albacete | C618G (TGC→CGC) | MEN 2A | 2 |
| Familia 5 | Albacete | M918T (ATG→ACG) | MEN 2B | 1 (de novo) |
| Familia 6 | Talavera | C620Y (TGC→TAC) | MEN 2A | 1 |
| Familia 7 | Mancha-Centro | C634Y (TGC→TAC) | MEN 2A | 1 |
| Familia 8 | Mancha-Centro | Sin datos | MEN 2A | 1 |
| Familia 9 | Ciudad Real | M918T (ATG→ACG) | MEN 2B | 1 (de novo) |
| Familia 10 | Cuenca | V804L (GTG→TTG) | MEN 2A | 2 |
| Familia 11 | Cuenca | Sin datos | MEN 2A | 4 |
| Familia 12 | Toledo | C620Y (TGC→TAC) | MEN 2A | 1 |
| Total | 20 | |||
MEN 2A: síndrome de neoplasia endocrina múltiple tipo 2A; MEN 2B: síndrome de neoplasia endocrina múltiple tipo 2B.
La edad al diagnóstico de los casos esporádicos, MEN 2A y MEN 2B fue de 54 (intervalo 14 a 82 años), 40 (intervalo 6-75 años) y 19 años (intervalo 18 a 21), respectivamente, siendo esta diferencia estadísticamente significativa (p<0,05). En el caso del grupo MEN 2A, al excluir del análisis al único paciente en edad pediátrica no se modifica la significación estadística entre los grupos; la edad media del grupo de MEN 2A una vez excluido el paciente de 6 años fue de 46 años (intervalo 21-75 años).
El motivo más frecuente que llevó al diagnóstico de CMT fue una palpación cervical patológica (nódulo tiroideo o adenopatía) en 41 pacientes (70,6%), y en segundo lugar, el estudio genético por ser familiar de una persona con CMT hereditario en 6 pacientes (10,3%). Otros motivos de diagnóstico fueron el hallazgo de un incidentaloma ecográfico (4 pacientes), el estudio de una metástasis a distancia (4 pacientes), el hallazgo incidental en la anatomía patológica de una tiroidectomía (2 pacientes) y el estudio de secreción ectópica de ACTH (un paciente).
Hallazgos analíticos, radiológicos y anatomopatológicosLa calcitonina sérica se determinó antes de la cirugía en 8 de los 58 pacientes, siendo 6 de ellos familiares de primer grado de un paciente diagnosticado de MEN. En cuanto a las pruebas de imagen al diagnóstico, la prueba más solicitada fue la ecografía (56 de 58 pacientes).
La presencia de multicentricidad en la pieza quirúrgica se observó en el 59% de los casos de MEN 2A analizados (10 de 17 pacientes, un paciente sin datos), el 50% de los casos de MEN 2B (uno de 2 pacientes) y en ningún caso de los esporádicos, siendo esta diferencia estadísticamente significativa (p<0,001).
EstadificaciónAl diagnóstico, 7 pacientes presentaban un estadio iv (2 con metástasis hepáticas, 2 con metástasis óseas, uno con metástasis pulmonares y 2 con enfermedad localmente avanzada), y de ellos, 6 correspondían a formas de CMT esporádicos y un paciente estaba asociado a MEN 2B. De los 20 pacientes con estadio iii, 15 correspondían a CMT esporádico, 4 pacientes estaban asociados a MEN 2A y un paciente estaba asociado a MEN 2B. De los 25 pacientes con estadios i y ii, 12 eran formas esporádicas y 13 estaban asociados a MEN 2A. No se disponía de los datos de 5 pacientes (tabla 3).
Estadio al diagnóstico
| Estadio (TNM) | n | CMT esporádico, n (%) | MEN 2A, n (%) | MEN 2B, n (%) |
| I | 15 | 7 (47) | 8 (53) | 0 |
| II | 10 | 5 (50) | 5 (50) | 0 |
| III | 20 | 16 (80) | 3 (15) | 1 (5) |
| IV | 7 | 6 (86) | 0 | 1 (14) |
| Sin datos | 6 | – | – | – |
MEN 2A: síndrome de neoplasia endocrina múltiple tipo 2A; MEN 2B: síndrome de neoplasia endocrina múltiple tipo 2B.
Fuente original de la clasificación TNM: AJJC Cancer Staging Manual, sexta edición (2002), publicado por Springer-Verlag, Inc., Nueva York.
Los 2 pacientes que no se intervinieron quirúrgicamente por enfermedad avanzada fallecieron antes de cumplirse un año del diagnóstico por causa del CMT. En el resto, el tratamiento fue quirúrgico, aunque no disponemos de datos sobre dicho procedimiento ni sobre la realización o no de vaciamiento ganglionar.
Tampoco disponemos de datos de seguimiento para 11 pacientes. Para los restantes 47, la mediana de seguimiento fue de 36 meses (RI 14 a 210 meses). De los 47 pacientes, 7 fallecieron como consecuencia del CMT (mediana de seguimiento de 68,5 meses, RI 24 a 152 meses); un paciente falleció por una causa no relacionada con el CMT; 29 pacientes estaban libres de enfermedad en el momento de la revisión (mediana de seguimiento de 36 meses, RI de 6 a 210 meses); 8 pacientes tenían enfermedad persistente sin metástasis a distancia (mediana de seguimiento de 33 meses, RI de 4 a 192 meses) y 4 pacientes tenían enfermedad persistente con metástasis a distancia (mediana de seguimiento de 45,5 meses, RI de 12 a 50 meses).
DiscusiónEn el presente estudio se confirman muchas de las características demográficas descritas en publicaciones previas de series de pacientes con CMT4. La edad media al diagnóstico, al igual que en la mayoría de los estudios publicados, se sitúa próxima a los 50 años de edad (51 años). Sin embargo, la proporción de mujeres (65%) es superior a la descrita previamente. Asimismo, se confirma la edad de diagnóstico más temprana en los casos hereditarios, probablemente debido a la detección precoz en los familiares de pacientes con MEN 2A, hecho que se asocia a un mejor pronóstico5. La inclusión de un único paciente en edad pediátrica (6 años) es posiblemente debida a la realización de tiroidectomía profiláctica en la mayoría de los casos pediátricos detectados gracias al estudio genético y, por lo tanto, no incluidos en este análisis. El CMT asociado a MEN 2B aparece a edades más tempranas y se asocia a una mayor agresividad y peor pronóstico6.
En cuanto a la prevalencia de casos hereditarios, ha resultado ser superior a la esperada (35%), si bien existe una amplia variabilidad en las publicaciones, pudiendo llegar hasta un 65% dependiendo de la región estudiada. La cercanía de la región de Murcia, donde ha sido descrita una mayor prevalencia de formas familiares, puede explicar la mayor presencia de formas familiares en el área de salud de Albacete (5 de las 11 familias de la Comunidad de Castilla-La Mancha)7.
Una vez detallado el tipo de mutación del protooncogén RET observamos que a nivel mundial la mutación más habitual es C634R, en el 52% de los casos, tal y como indica el International RET Mutation Consortium Study8. Sin embargo, la mutación más habitual en España es C634Y, como ha sido descrito en el estudio de Sánchez et al. que identifica que esta mutación abarca el 73% de los casos9. En este sentido, los estudios regionales realizados en España apoyan este hallazgo, como es el caso de la población murciana, con una prevalencia publicada de hasta el 80% de dicha mutación10. Esto planteó la hipótesis de un antepasado común para explicar la mayoría de los casos de la población española, que sin embargo, en el caso de Castilla-La Mancha explicaría solo 4 casos de 15 y representa 3 familias de 9. No deja de ser llamativo este hecho, ya que en el área sanitaria de Castilla-La Mancha, aunque la mutación mayoritaria es la C634Y, solo representa un 26% de los casos, en lugar del 73% publicado para la población española9, observándose en el presente estudio una importante variabilidad del tipo de mutación del protooncogén RET, que aparece como excepción comparada con la del resto de España.
En cuanto a la multicentricidad del tumor, en nuestra serie es un dato patognomónico de CMT hereditario. Sin embargo, los datos publicados hasta la fecha determinan la presencia de múltiples focos de CMT también en casos esporádicos, siendo su proporción menor comparada con casos familiares, pero variable según las series hasta un 53% de los casos esporádicos10. Por todo ello sería necesario homogeneizar el criterio diagnóstico de multicentricidad en nuestra área de cara a conseguir un diagnóstico histológico más preciso.
Al analizar el estadio al diagnóstico en nuestra serie, aproximadamente la mitad de los pacientes presentaba un estadio avanzado (iii o iv) acorde con los datos de otras series4,11,12, lo que conlleva un peor pronóstico13. En este sentido, la medición rutinaria de calcitonina en el manejo del nódulo tiroideo es controvertida2. Se midieron los niveles de calcitonina al diagnóstico en 8 de 58 pacientes, por ser familiares de portadores de CMT hereditario o por un diagnóstico citológico de CMT. Por lo tanto, podemos afirmar que para el manejo del nódulo tiroideo en nuestra área no se solicita la medición de calcitonina de manera rutinaria, mientras que sí se basa en la ecografía tiroidea (solo 2 de nuestros pacientes no la tenían). Aunque en los últimos años los datos sobre la medición rutinaria de la calcitonina parecen indicar que sería una medida coste-efectiva, sigue sin existir un acuerdo unánime sobre su detección sistemática en el estudio del nódulo tiroideo. Concretamente, mientras el consenso europeo sobre manejo del cáncer diferenciado de tiroides se pronuncia a favor14 y la guía de la American Thyroid Association (ATA) sobre manejo del nódulo tiroideo y cáncer diferenciado de tiroides2 no se pronuncia ni a favor ni en contra, la guía de la American Thyroid Association sobre manejo del cáncer medular de tiroides15 recomienda no medir la calcitonina de manera rutinaria. Sería interesante la realización de un estudio de coste-efectividad en nuestro medio para definir mejor la utilidad de esta prueba en el estudio inicial del nódulo tiroideo.
En cuanto a los posibles sesgos del estudio, debemos destacar el diseño, ya que al ser un análisis retrospectivo no se garantiza la inclusión de todos los casos de CMT de Castilla-La Mancha, lo que explicaría la disparidad de la prevalencia por regiones. Otra explicación de dicha variabilidad puede ser la cercanía de la Comunidad de Madrid, debido a la posible captación de pacientes de provincias limítrofes por su condición clásica de centro de referencia, que podría explicar la baja prevalencia de CMT en las provincias de Toledo y Guadalajara. Por otra parte, la cercanía del área de Albacete a la Comunidad de Murcia puede justificar la mayor prevalencia de formas familiares en este área de salud.
Otra limitación del estudio reside en la pérdida de seguimiento de varios pacientes, que resta precisión a los datos presentados.
En conclusión, diremos que el manejo diagnóstico del nódulo tiroideo en nuestro medio se basa en la ecografía cervical sin incluir calcitonina de manera rutinaria. En esta serie destacan la alta proporción de casos familiares, la variabilidad del genotipo del protooncogén RET con respecto al resto de la población española, y la heterogeneidad de la prevalencia del CMT en las distintas áreas sanitarias. El estadio menos avanzado de los casos familiares, excluyendo el MEN 2B, nos recuerda la importancia del diagnóstico precoz y la necesidad de optimizar este aspecto para mejorar el pronóstico del CMT.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
