



La sarcoidosis (SA) es una enfermedad sistémica de etiología desconocida caracterizada por la presencia histológica de granulomas no caseificados1. Su incidencia en España es baja, afectando a 1,2–1,5 personas por cada 100.000 habitantes. Los pulmones, la piel, los ojos y los ganglios linfáticos son los órganos más frecuentemente afectados, mientras que solo el 5% de los pacientes presenta manifestaciones neurológicas2. Aunque la sospecha de neurosarcoidosis (NSA) debe existir en todo paciente con SA conocida que desarrolla sintomatología neurológica, el diagnóstico es realmente difícil cuando esta es la clínica de debut. Presentamos el caso de una paciente que consultó por sintomatología compatible con diabetes insípida (DI) central, que resultó ser secundaria a NSA.
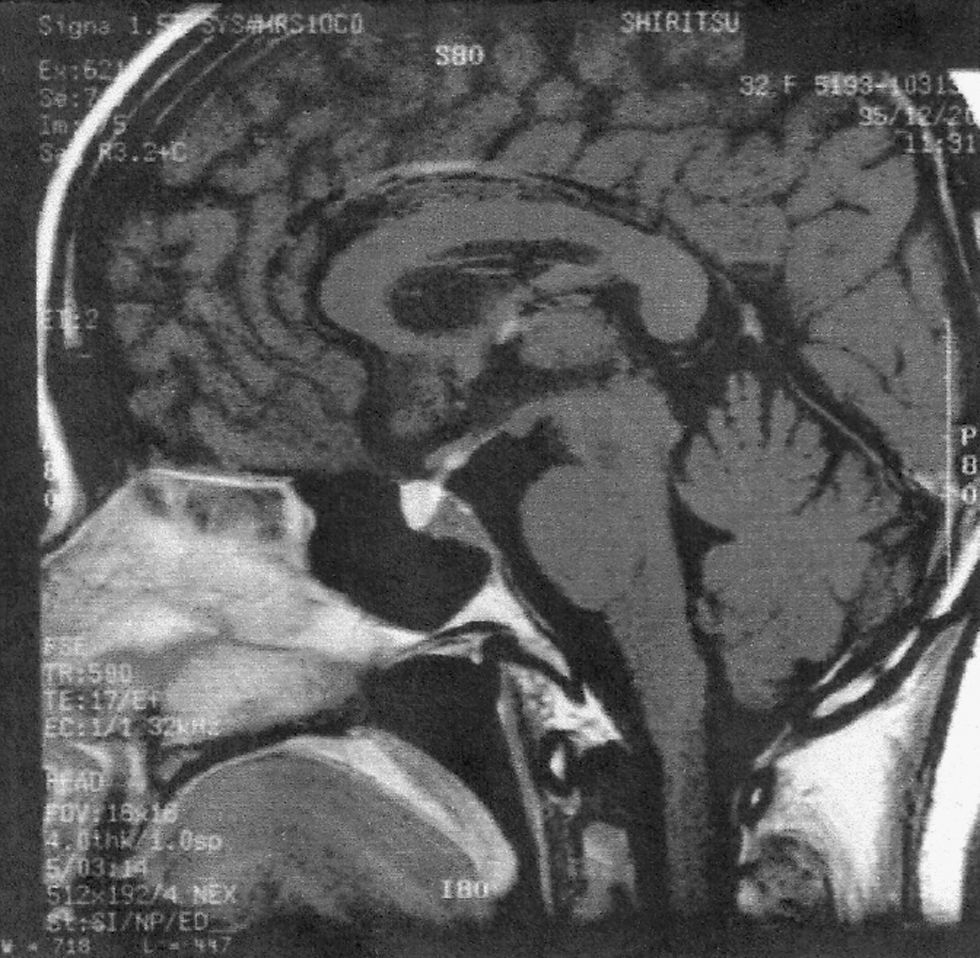
Mujer de 40 años, sin antecedentes de interés, que consultó por un cuadro de poliuria de más de 3l/d con hipodipsia. La paciente no refería otra sintomatología de interés, salvo la presencia de cefaleas ocasionales que atribuía a disminución de la agudeza visual. A la exploración física, únicamente destacaba la presencia de una pupila izquierda midriática con discreta ptosis palpebral, que resultó ser consecuencia de la afectación del iii par craneal. La analítica de sangre mostró un sodio de 150mEq/l (rango de referencia: 135–145) con osmolaridad plasmática de 292mOsm/kg. Los niveles de glucosa, calcio, potasio y demás parámetros bioquímicos fueron normales. La analítica de orina mostró una osmolaridad de 200mOsm/kg. Sospechando una DI como responsable del cuadro, se realizó el test de deshidratación de Miller. Este confirmó el diagnóstico de DI central, por lo que se inició tratamiento con desmopresina intranasal. La función basal hipofisaria resultó normal (tabla 1). La tomografía computarizada (TC) craneal mostró una masa paraselar de 2,5cm realzada con contraste, sospechosa de aneurisma del polígono de Willis, pero la angiografía cerebral fue normal. La resonancia magnética (fig. 1) evidenció dicha lesión selar-paraselar con afectación hipotalámica y, además, una pequeña lesión frontal y en el cráneo. La radiografía craneal mostró múltiples lesiones líticas y la misma lesión ocupante de espacio, que fueron biopsiadas. La biopsia demostró la existencia de inflamación granulomatosa sin necrosis caseosa, con sospecha principal de SA o menos probable histiocitosis X o tuberculosis. El cultivo, la tinción y la reacción en cadena de la polimerasa frente a Mycobacterium tuberculosis fueron negativas y una nueva revisión histológica descartó la posibilidad de histiocitosis. Los niveles de enzima convertidora de angiotensina plasmática fueron normales (50U/l; rango de referencia: 18–55U/l). La radiografía de tórax y la TC torácica mostraron adenopatías hiliares bilaterales compatibles con SA, aunque la biopsia transbronquial no pudo evidenciar granulomas. Con el diagnóstico de SA con afectación del sistema nervioso, se inició tratamiento con 60mg/día de prednisolona en pauta descendente. Cuando la dosis fue inferior a 30mg/d la paciente desarrolló neuralgia del trigémino, por lo que se añadió tratamiento con hidroxicloroquina. La oftalmopatía y la neuralgia desaparecieron, con mejoría en las pruebas de imagen. Seis años después, la paciente continúa con glucocorticoides de mantenimiento y la hidroxicloroquina se ha sustituido por metotrexato, por la aparición de efectos adversos. La TC craneal se mantiene estable y el mal control de la DI constituye el problema principal, requiriendo varios ingresos para ajuste de la dosis de desmopresina.
Estudio de la función basal hipofisaria
| Hormona (rango de referencia) | Niveles |
| Tirotropina (0,35–5,5mU/l) | 2,5 |
| Tiroxina libre (0,9–1,8ng/dl) | 1,4 |
| Triyodotironina libre (2,3–4,3pg/ml) | 3,1 |
| Hormona adrenocorticotropa (10–80ng/l) | 25 |
| Cortisol (6,5–22,5ng/dl) | 19 |
| Hormona de crecimiento (0–10ng/ml) | 1,9 |
| Factor de crecimiento insulínico tipo 1 (105–353ng/ml) | 203 |
| Hormona foliculoestimulante (8–33mU/ml) | 15 |
| Hormona luteinizante (20–80mU/ml) | 32 |
| Estradiol (1332–5290pg/ml) | 1.650 |
| Prolactina (3–31ng/ml) | 12 |

La SA puede afectar a cualquier área del sistema nervioso central o periférico. Lo más frecuente es la afectación del vii par craneal3, aunque otras manifestaciones incluyen meningitis asépticas, efecto masa, vértigo o hidrocefalia4. La disfunción endocrina por infiltración hipotalamicohipofisaria ocurre en el 10–25% de los pacientes con NSA, siendo el hipogonadismo y las alteraciones del balance hídrico las más frecuentes5. El 38,5% de los pacientes presenta hipogonadismo hipogonadotropo por alteraciones en la liberación de la hormona liberadora de gonadotropinas, mientras que en un 9% de los casos el hipogonadismo es secundario a hiperprolactinemia y se acompaña de galactorrea. La DI central es la alteración del balance hídrico más habitual (37%), si bien existen casos de síndrome de secreción inadecuada de vasopresina. Además, un 23% de los pacientes con afectación hipotalamicohipofisaria presenta panhipopituitarismo, y aunque también se han descrito casos de disfunción hormonal hipofisaria aislada, estos son mucho menos frecuentes (<5%). Todas estas alteraciones hormonales se deben principalmente a la infiltración granulomatosa a nivel hipotalámico, aunque esta también puede ocurrir en el tallo hipofisario o en la hipófisis. En este último caso, el diagnóstico diferencial es más complicado, debiendo distinguirse de otras causas de masa selar.
Los pacientes se clasifican en aquéllos con NSA posible, probable o definitiva, basándose en los criterios establecidos por Zajicek en 19996. El diagnóstico definitivo se realiza tras la exclusión de otras enfermedades a las que simula (infecciones —principalmente tuberculosis—, histiocitosis, linfoma, esclerosis múltiple, vasculitis, etc.) y la demostración histológica de granulomas no caseificados, por lo que frecuentemente se requiere biopsia cerebral. La resonancia magnética es la técnica de imagen más sensible y específica para el diagnóstico, aunque es normal en el 10% de los pacientes6. La administración de contraste permite observar realce a nivel leptomeníngeo, anomalías parenquimatosas y, ocasionalmente, lesiones a nivel de los pares craneales9. La presencia de lesiones líticas en el cráneo, como en este caso, es excepcional dado que en caso de afectación ósea (el 1–34% de pacientes), esta suele limitarse a falanges y metacarpo/metatarso. Así, en caso de afectación del esqueleto axial y/o el cráneo debe realizarse el diagnóstico diferencial con otras causas más frecuentes, como mieloma múltiple, linfoma, metástasis, enfermedad de Paget, histiocitosis X o tuberculosis.
Los objetivos del tratamiento son disminuir la sintomatología y la isquemia ocasionada por la inflamación perivascular, ya que no se ha demostrado que modifique la historia natural de la enfermedad7. Aunque no disponemos de ensayos clínicos para determinar el manejo más adecuado, este incluye glucocorticoides, inmunosupresores, inmunomoduladores o, si fracasan todas las medidas anteriores y con resultados variables, la radioterapia8. Los glucocorticoides a altas dosis (prednisolona de 1mg/kg/d) constituyen el tratamiento de elección. Tras 4–6 semanas, empiezan a disminuirse progresivamente, aunque la mayoría de los pacientes requieren dosis de mantenimiento de 10–15mg/d para evitar recaídas. En pacientes con afectación neurológica rápidamente progresiva y grave, deben administrarse 20mg/kg/d de metilprednisolona intravenosos durante 3 días, seguido de altas dosis de prednisolona. En caso de enfermedad refractaria o necesidad de dosis elevadas de glucocorticoides, se emplean inmunosupresores en adyuvancia, principalmente ciclosporina A y metotrexato. Ningún agente parece más efectivo que otro y, dado que la respuesta es variable y el beneficio modesto, habitualmente no puede suspenderse el tratamiento glucocorticoideo. En adición, inmunomoduladores como la hidroxicloroquina se utilizaron con buena respuesta. Además, recientemente se han ubicado 5 casos de NSA refractaria a los anteriores, tratados con infliximab con muy buenos y prometedores resultados9.
El pronóstico generalmente es bueno, aunque los pacientes con afectación del parénquima cerebral tienen una evolución progresiva o con frecuentes recaídas, y la presencia de hidrocefalia implica una mortalidad de hasta el 75%10. La afectación endocrinológica habitualmente no mejora con el tratamiento, por lo que precisa tratamiento hormonal sustitutivo de por vida.
En conclusión, la SA es una enfermedad sistémica poco frecuente que rara vez afecta al sistema nervioso; si bien, cuando lo hace, puede simular múltiples enfermedades neurológicas, por lo que el diagnóstico supone realmente un reto. El estudio completo de la función hipotalamicohipofisaria es fundamental para identificar déficits hormonales o alteraciones del balance hídrico. Asimismo, un diagnóstico precoz con un tratamiento agresivo es preciso para lograr resultados satisfactorios, aunque las endocrinopatías suelen persistir y precisar tratamiento hormonal sustitutivo crónico.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
