


There is a growing perception that profound environmental changes in areas such as diet and lifestyle that began with introduction of agriculture and taming of animals about 10,000 years ago actually occurred very recently on an evolutionary time scale. Evolution from the paleolithic diet to our current modern intake pattern also resulted in several changes in feeding behavior, including increased consumption of processed foods rich in sodium and hydrogenated fats and low in fiber.1–3 Changes in several food parameters, such as increased calorie intake (mainly of foods rich in saturated/trans fatty acids) and serving sizes, including introduction of “super size” meals, are known to play roles in the advent of non-communicable chronic diseases.4,5
The interest in the study of lipids has increased since the 19th century. Vogel, in 1847, was the first researcher to detect the presence of cholesterol in atherosclerotic plaques. A century later, Bang & Dyerberg6 noted that Eskimos had a low incidence of cardiovascular diseases despite their high-fat diet, and suggested for the first time that omega-3 fatty acids are responsible for inhibiting obesity-related diseases.
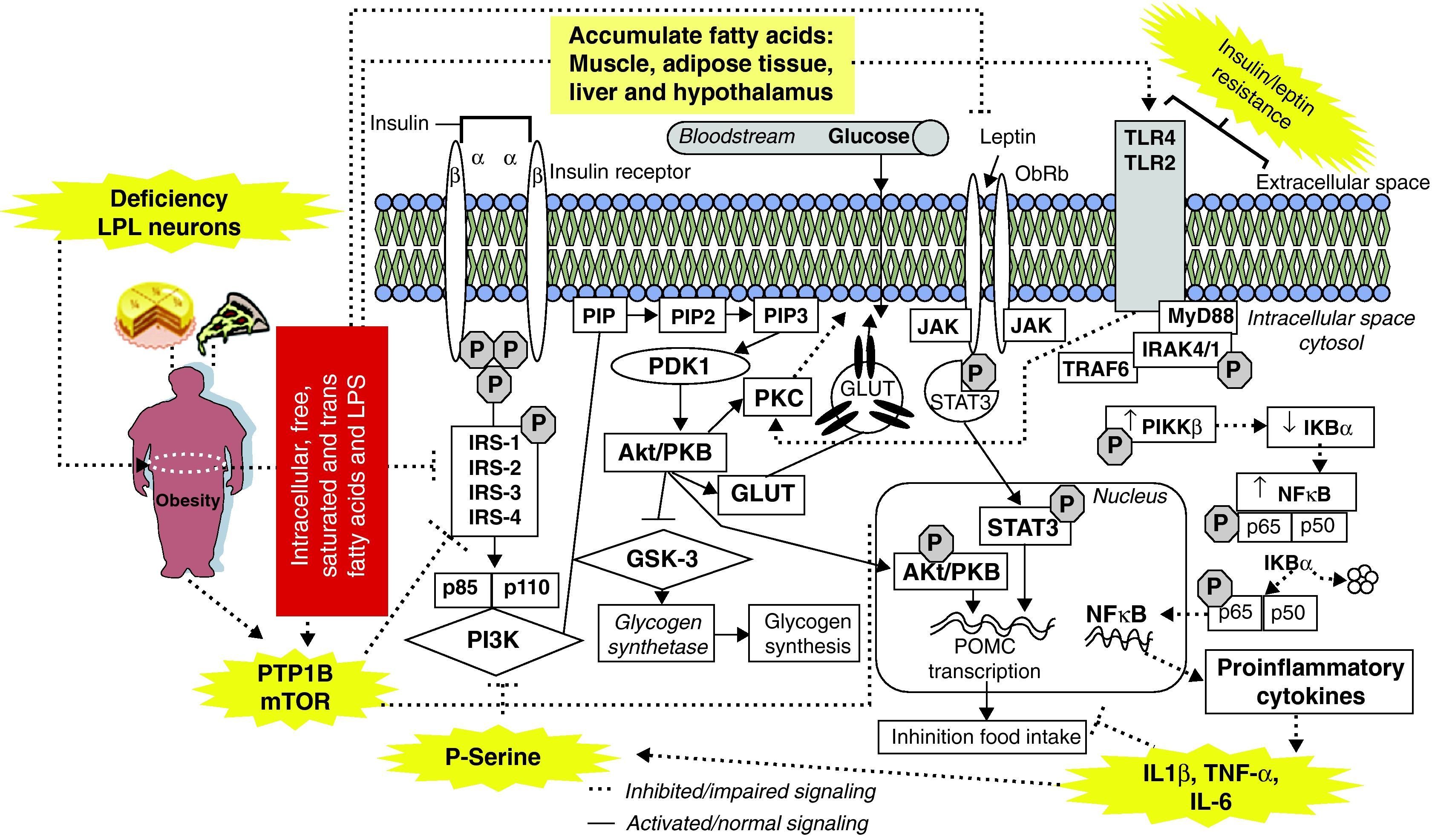
After World War II and industrialization, fast-food chains began providing meals rich in saturated/trans fatty acids, which may be responsible for the high prevalence of obesity.7 Thus, in 1994, after the discovery of leptin,8 several studies demonstrated that high-fat diets lead to obesity and induce inflammation in peripheral tissues and hypothalamus, promoting insulin and leptin resistance.9,10 These results may partially be explained by accumulation of intracellular fatty acids (e.g. acyl-CoA); free fatty acids (FFAs) released by adipocytes via lipolysis may induce inflammatory changes and lead to impaired insulin and leptin signaling.11 Recently, Wang et al.12 demonstrated that lipoprotein lipase (LPL), a serine hydrolase that releases FFAs from circulating triacylglycerol-rich lipoproteins, might contribute to FFA-mediated signalling in the central nervous system. The authors found that neuron-specific homozygous mutant (NEXLPL-/-) mice are hyperphagic and become obese by 16 weeks of age (fig. 1).

Obesity and saturated/trans fatty acids-rich diets leads to insulin/leptin resistance and inflammation in several tissues.
IRS-1 and 2: insulin receptor substrate 1 and 2, IKK-β: inhibitor of nuclear factor-κB kinase, p85: regulatory subunit of PI3-K, p110: catalytic subunit of PI3-K, PI3-K: phosphatidylinositol 3-kinase, PIP: phosphatidylinositol protein, PIP2: phosphatidylinositol-4,5-bisphosphate, PIP3: phosphatidylinositol-3,4,5-triphosphate, PDK1: 3-phosphoinositide–dependent protein kinase, Akt/PKB: protein kinase B, PKC: protein kinase C, GSK-3: glycogen synthase kinase-3, GLUT-2,4: glucose transporter 2,4, MyD88: myeloid differentiation factor 88, p50: p50 subunit of NFκB, p65: p65 subunit of NFκB, pIKBα: phospho-inhibitory subunit of NF-κBα, pIKKβ: phosphorylated IKKβ, NFκB: nuclear factor kappa B, IL-6: interleukin 6, IL-8: interleukin 8, IL-1β: interleukin-1β, TNF-α: tumor necrosis factor alpha, SOCS3: suppressor of cytokine signaling-3, TLR4: toll-like receptors, LPL: lipoprotein lipase, LPS: lipopolysaccharides, JAK: Janus kinase, STAT3: signal transducer and activator of transcription 3.
These increased acyl-CoA/FFA levels activate the serine/threonine kinase protein kinase C (PKC), which can phosphorylate insulin receptor substrates (IRSs) and activate toll-like receptors (TLR2/4), activating the nuclear factor-κB kinase complex (IKK-β) and nuclear factor kappa B (NF-κB P50/p65), and increasing secretion of inflammatory cytokines leading to insulin and leptin resistance. Moreover, saturated/trans fatty acids and enteric lipopolysaccharides (LPS) may act as direct ligands for TLR2/4,9,11,13,14 activating PKC and resulting in insulin resistance (fig. 1). However, other signaling pathways that are involved in insulin resistance can also be regulated by saturated/trans fatty acids through TLR4; these include the mitogen-activated protein kinases (MAPKs), c-Jun amino-terminal kinase (JNK), endoplasmic reticulum (ER) stress, the unfolded protein response (UPR) and ceramide pathways, as well as inhibition of peroxisome proliferator-activated receptor (PPAR) gamma and adiponectin signaling.10,14–16 Consumption of saturated fatty acid-rich diets blunts leptin anorexigenic signaling in hypothalamus through an inflammation-dependent mechanism, probably by activation of TLR4 and myeloid differentiation factor 88 (MyD88)17,18 (fig. 1).
Another potential cause for obesity-associated diseases is activation of protein-tyrosine phosphatase (PTP)-1B, a key enzyme involved in regulation of reversible tyrosine phosphorylation. PTP-1B may be activated by saturated fatty acids and inflammation and, once activated, causes inactivation of insulin receptor by removing phosphates from the active insulin receptor and its substrates.19,20 Interestingly, PTP-1B may also dephosphorylate Janus kinase/signal transducer and activator of transcription (JAK/STAT3), decreasing leptin effects and causing leptin resistance.20–22 Moreover, studies show that stimulation of the mammalian target of the rapamycin (mTOR/p70S6K) pathway is associated with IRS-1/2 serine phosphorylation and impaired Akt activation by insulin23,24 (fig. 1).
In summary, it is evident that changes in diet and advent of industrialization, with the rise of fast food chains and sedentary lifestyles, have increased the prevalence of low-grade inflammatory conditions such as cardiovascular diseases, metabolic syndrome, diabetes, and obesity. However, subjects who have not made these dietary changes do not develop metabolic abnormalities. In addition, healthy eating, with low saturated/trans fatty acid or high polyunsaturated fatty acid levels, may partially reverse these diseases. Thus, future studies should be performed to evaluate the consequences of this change in diet; such studies should particularly focus on as yet uncharacterized aspects of fat intake and on anti-inflammatory therapies that may be used for the treatment of obesity-related diseases.
