


La displasia ósea fibrosa es un trastorno esquelético originado por una mutación del gen GNAS1 con un amplio espectro de presentaciones clínicas. Hay formas monostóticas y poliostóticas puras y relacionadas con trastornos extraesqueléticos (síndrome de McCune-Albright). La morbilidad está determinada por el desarrollo de deformidades, fracturas y dolor óseo y, en ocasiones, compresión de nervios craneales y degeneración sarcomatosa. La cirugía traumatológica representa el abordaje principal de las lesiones complicadas y actualmente las posibilidades de tratamientos farmacológicos eficaces son bastante limitadas. La administración de bisfosfonatos intravenosos en ciclos es la intervención más justificada para la mejoría sintomática y la disminución del riesgo de fracturas, aunque son necesarios estudios clínicos controlados que evalúen la eficacia y la seguridad de estos tratamientos. Presentamos un caso clínico de displasia ósea fibrosa poliostótica en un varón de 19 años.
Fibrous dysplasia of bone is a skeletal disease due to a specific mutation in the GNAS1 gene with a broad spectrum of clinical presentations ranging from isolated monostotic and polyostotic forms to associations with other extra-skeletal manifestations (McCune-Albright syndrome). The main complications are bone pain, deformities and fractures and sometimes cranial nerve compression and the development of bone sarcomas.
Traumatologic surgery represents the principal approach in complicated lesions. Currently, the efficacy of pharmacological therapies is fairly limited. Cyclic administration of intravenous biphosphonates is the most effective approach to achieve symptomatic improvement and to lower the risk of fractures, although more evidence from well-designed randomized clinical trials evaluating the safety and efficacy of these treatments is still required. We present a case of polyostotic fibrous dysplasia of bone in a 19-year-old male.
La displasia fibrosa ósea (DF) es un trastorno esquelético originado por una mutación genética específica con un amplio espectro de presentaciones clínicas que van desde hallazgos radiológicos en pacientes asintomáticos hasta anomalías óseas severas de carácter grave e incapacitante1. La enfermedad puede afectar a un solo hueso (DF monostótica), a varios (DF poliostótica) o al esqueleto entero (DF panostótica). En ocasiones, las lesiones óseas características de la enfermedad pueden tener relación con alteraciones extraesqueléticas, como hiperpigmentación cutánea, hiperfunción de glándulas endocrinas, tubulopatía renal o mixomas en distintos órganos.
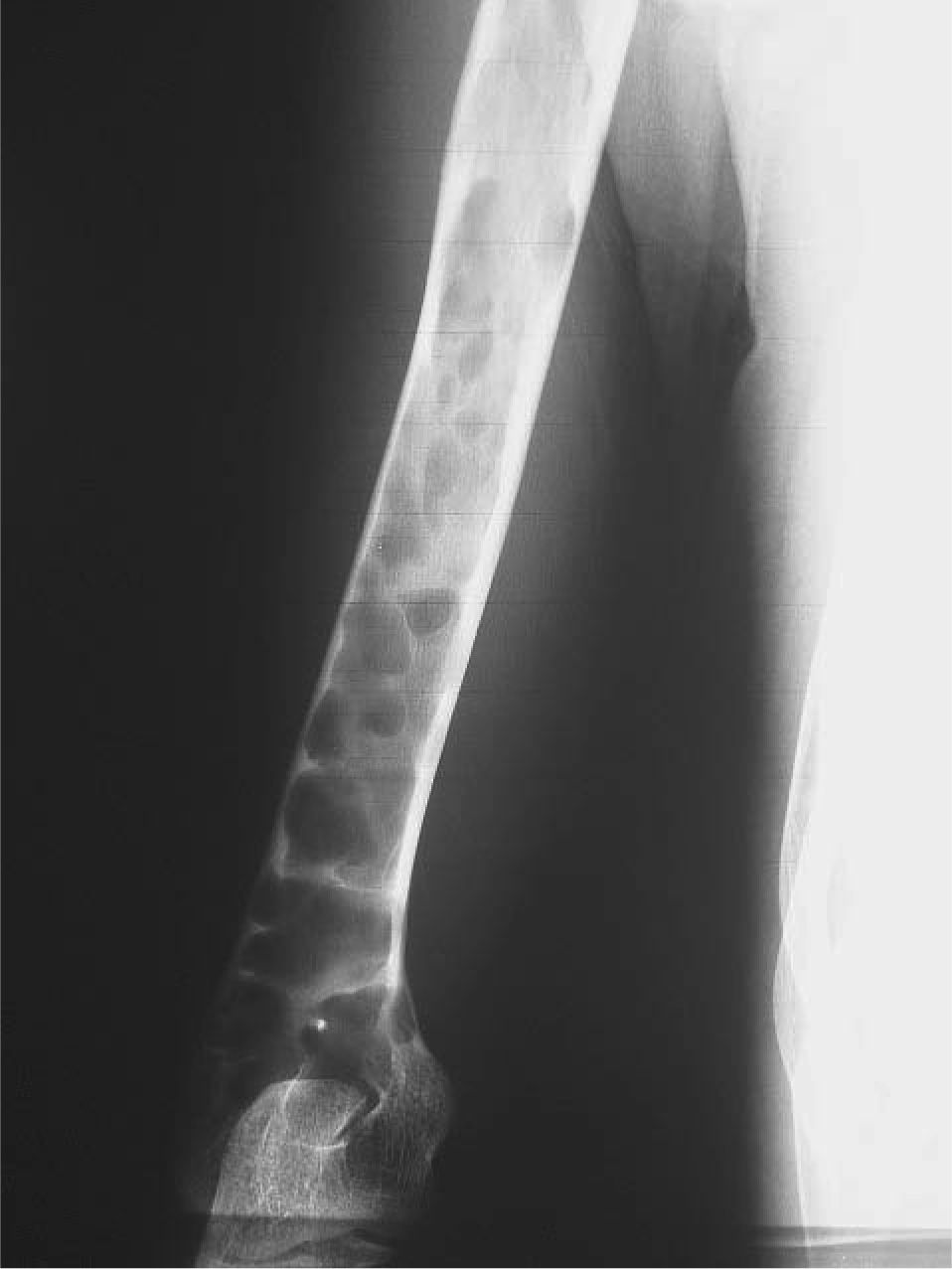
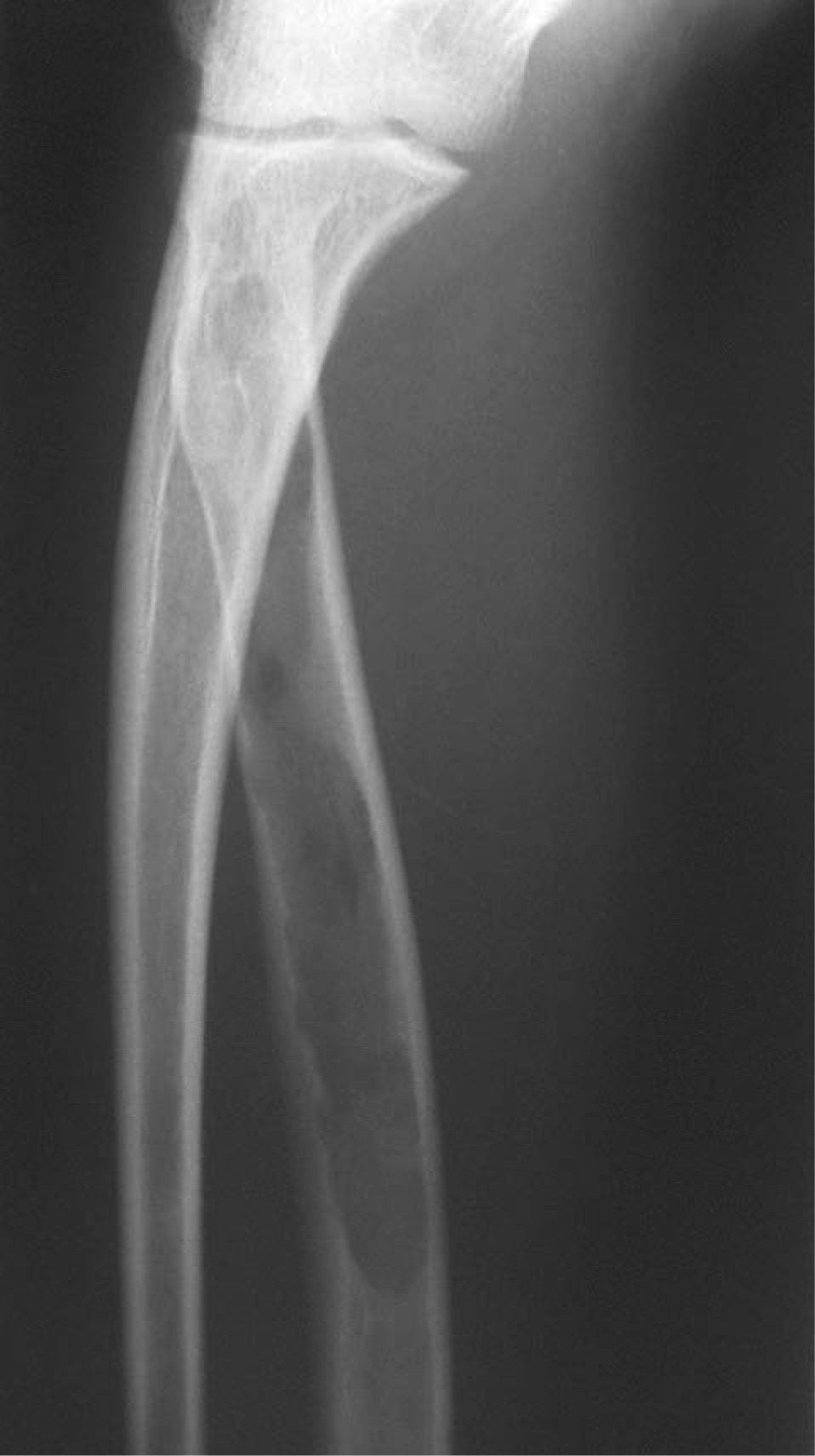
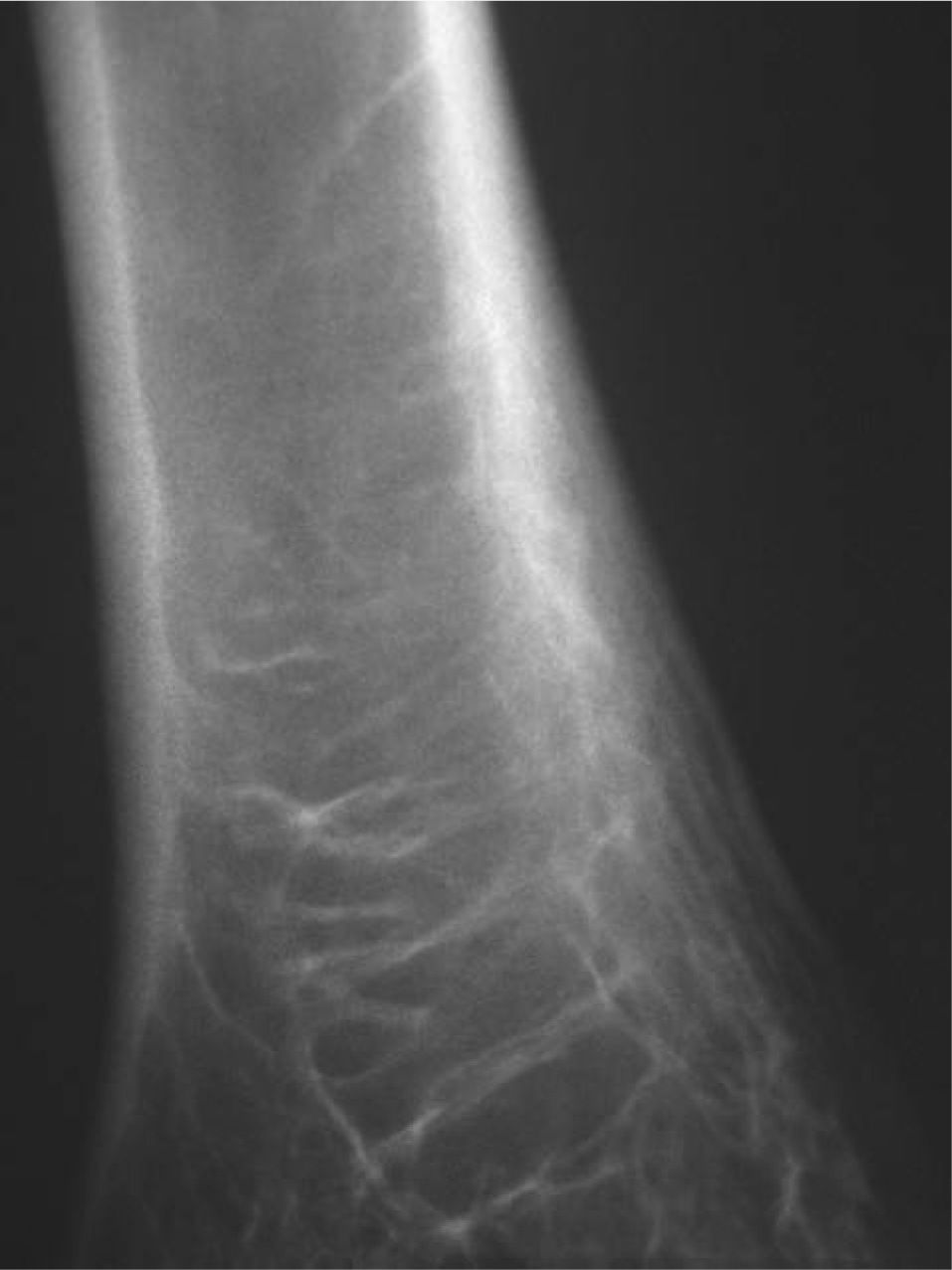
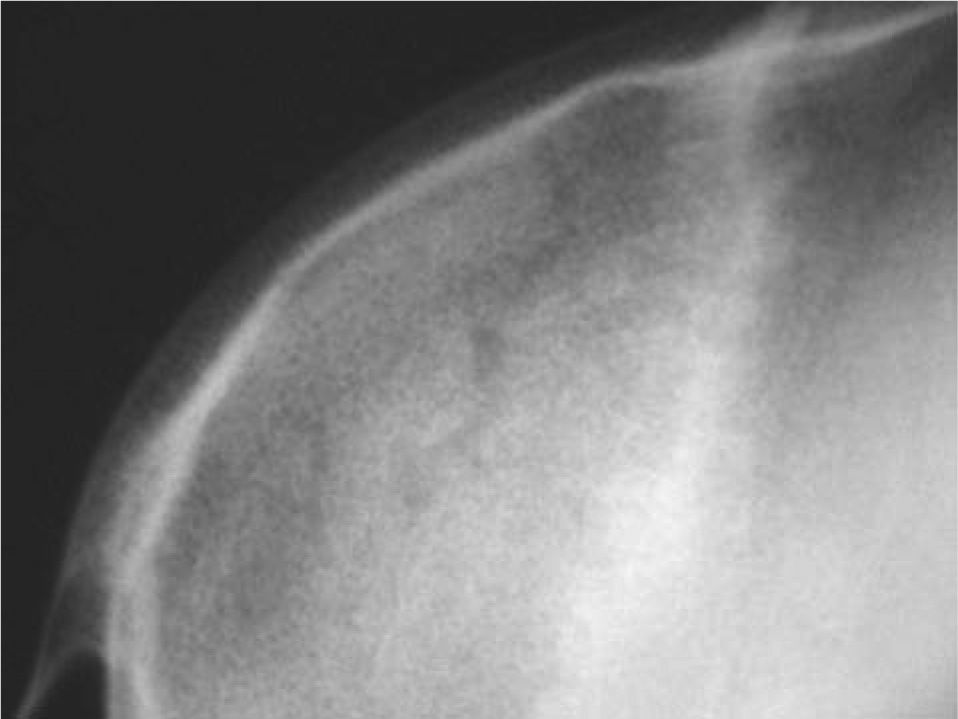
CASO CLÍNICOPaciente de 19 años remitido a la unidad de metabolismo óseo tras ser asistido en traumatología por presentar múltiples fracturas óseas ante traumatismos menores. En el momento de la valoración inicial había padecido 4 fracturas de radio izquierdo y durante los siguientes 6 meses presentó una nueva fractura en esa zona y otra en el radio derecho. Tratado mediante inmovilización con escayola e injerto óseo en el radio izquierdo tras la última fractura. No presentaba antecedentes familiares de enfermedades óseas o fracturas por fragilidad ni otros antecedentes personales patológicos de interés. Sin antecedentes de pubertad precoz. En la exploración física no se evidenciaron lesiones cutáneas hiperpigmentadas, signos clínicos de hipersecreción de glucocorticoides, hormonas tiroideas o somatotrofina ni disfunción de nervios craneales. La densitometría ósea (DXA, Hologic QDR 4500w) evidenció osteopenia en la zona lumbar (Z-score, –1,7 desviaciones estándar [DE]) y densidad mineral ósea normal en fémur proximal (Z-score del fémur total, –0,1 DE; cuello femoral, –0,6 DE). La analítica mostraba concentraciones normales de hormonas tiroideas, cortisol urinario de 24 h, IGF-I y testosterona; calcemia, 10,1 mg/dl; fosfatemia, 5,3 mg/dl; PTHi, 26,9 pg/ml (valores normales [VN], 15-65); 25(OH)vitamina D, 43,8 ng/ml (VN, 9-34); osteocalcina, 51,4 ng/l (VN, 12-52); TRALP-5b, 5,1 U/l (VN, 1,03-4,15); osteoprotegerina, 2,9 pg/ml (VN, 4,1 ± 0,33); isoenzima ósea de la fosfatasa alcalina (bALP), 36,6 ^g/ml (VN, 7,43-31,37); CTx, 2.370 ng/ml (VN, 0,158-0,442). La bioquímica general y el hemograma fueron normales. El estudio radiológico evidenció múltiples lesiones quísticas expansivas en las metáfisis de ambos radios y región metafisodiafisaria de ambos húmeros, con adelgazamiento de la cortical y aspecto en "cristal esmerilado o deslustrado" (figs. 1-3). Calota craneal con aspecto "en sal y pimienta" (fig. 4). Las extremidades inferiores, manos, el tórax y la columna sin evidencias de lesiones óseas. Con los datos clínicos y radiológicos expuestos se asumió el diagnóstico de displasia fibrosa poliostótica.
DISCUSIONEtiopatogenia



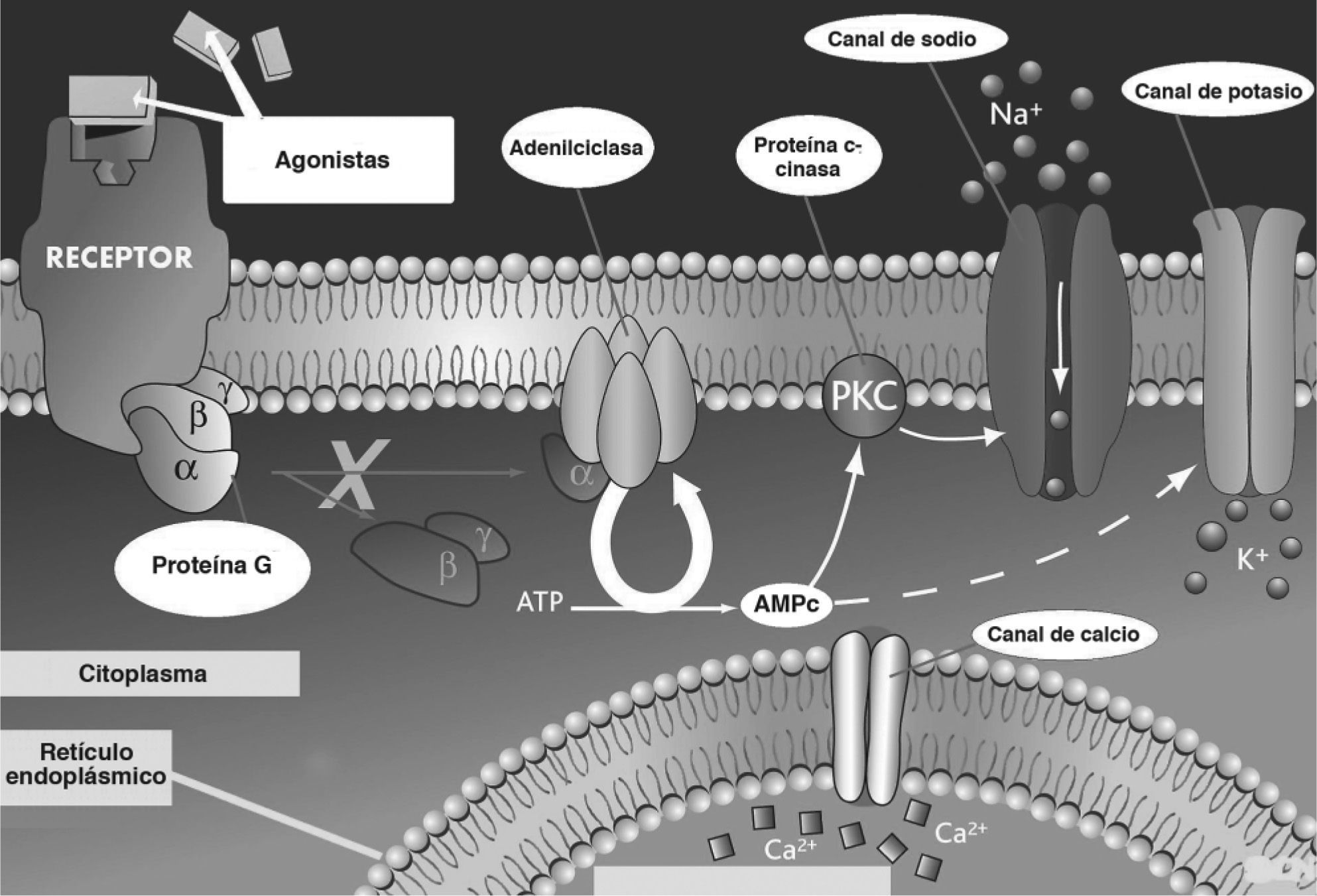
La anomalía subyacente a las distintas formas de DF es una mutación activadora del gen GNAS1, que codifica la subunidad alfa de la proteína G activadora (Gsa)2 (fig. 5). Estas mutaciones no son de carácter hereditario, sino que se producen tras la fase cigótica del desarrollo y originan un mosaicismo somático, en el cual coexisten líneas celulares con la alteración genética y poblaciones celulares normales. El momento del desarrollo prenatal en el cual se produce la mutación y la extensión y la viabilidad del clon resultante (el conjunto de células derivadas de la célula con la mutación original) son los factores determinantes de la distribución variable posnatal de células mutadas en el organismo y, por lo tanto, de la gravedad y la extensión de la enfermedad. Como consecuencia de esta mutación se produce una anormal activación constitutiva de la adenilciclasa asociada a la proteína G y el exceso de adenosinmonofosfato cíclico (AMPc) resultante actúa como segundo mensajero intracelular mediando los efectos patológicos en las células afectadas. En el tejido óseo, esta anomalía induce una expansión del conjunto de células osteoprogenitoras con acumulación progresiva en los espacios medulares que produce una pérdida localizada de tejido hematopoyético y fibrosis medular. Las células osteogénicas mutadas son funcional y morfológicamente anómalas, y originan una deposición de tejido óseo defectuoso. Así, las trabéculas óseas presentan alteraciones estructurales (patrón en "escritura china" o "sopa de letras") con importantes defectos de mineralización y distensión anómala3. Además, el colágeno del osteoide presenta alteraciones de orientación y composición bioquímica. El tejido displásico es altamente vascularizado y, por este motivo, es propenso a sangrados espontáneos con formación de quistes poshemorrágicos. En casos extremos, en formas de DF poliostótica se producen cortocircuitos arteriovenosos que pueden conducir al desarrollo de una insuficiencia cardíaca con alto gasto. Aunque no son necesarios para el diagnóstico, los valores de los marcadores bioquímicos de remodelado óseo suelen estar elevados, como en el caso presentado, en que se evidenciaron cifras incrementadas de marcadores de reabsorción ósea.
Manifestaciones clínicas
Las alteraciones fisiopatológicas descritas son más pronunciadas y evidentes durante la fase de crecimiento óseo rápido, de modo que la presentación clínica más frecuente se observa en la infancia y la adolescencia. Sin embargo, es infrecuente la aparición de alteraciones esqueléticas durante la lactancia y, en tales casos, conlleva una alteración grave y extensa con múltiples complicaciones. Las manifestaciones clínicas más frecuentes al inicio son el dolor óseo, las deformidades y las fracturas. El dolor afecta usualmente a costillas, huesos largos y craneofaciales, suele ser intenso y requerir la administración de analgésicos opioides. Las lesiones que afectan a la columna y la pelvis conllevan dolor de menor intensidad1.
El hallazgo de lesiones cutáneas hiperpigmentadas de tipo "café con leche" o disfunciones endocrinas concomitantes debe orientar al diagnóstico de síndrome de McCune-Albright. Las lesiones de la piel suelen observarse en niños y típicamente afectan al rostro, el tórax y las extremidades con tendencia a seguir una distribución en torno a la línea media y líneas de desarrollo de Blashko4.
Las deformidades de los huesos de las extremidades se producen como consecuencia de la extensión del tejido fibrótico, que presenta una resistencia biomecánica disminuida, las secuelas acumuladas de las fracturas y la formación de lesiones quísticas. Las fracturas pueden aparecer en cualquier hueso afectado, en forma espontánea o ante traumatismos menores, y afectan con frecuencia a la metáfisis proximal del fémur y los huesos de la base del cráneo5.
Hay casos en que la DF se relaciona con otras alteraciones extraesqueléticas que pueden modificar la expresión clínica de la enfermedad. La tubulopatía con pérdida renal de fosfatos e hipofosfatemia interfiere en la mineralización ósea acentuando las deformidades y fragilidad de los huesos afectados6. Asimismo, algunas disfunciones endocrinas que conforman el cuadro clínico del síndrome de McCune-Albright (pubertad precoz, exceso de hormona de crecimiento, hipertiroidismo, hipercortisolismo) pueden afectar de modo significativo la expresión de las anomalías esqueléticas7.
En raras ocasiones, se ha descrito malignización de las lesiones de DF (menos del 1%), y el sarcoma osteogénico es la variante más frecuente en estos casos8. El tratamiento previo con radioterapia externa es un claro factor de riesgo y el curso clínico del tumor es agresivo y de mal pronóstico9.
Características radiológicasLos hallazgos radiológicos son característicos, aunque variables y de carácter evolutivo. En niños, las imágenes habitualmente observadas corresponden a lesiones medulares expansivas con adelgazamiento de la cortical y aspecto en "cristal esmerilado". Cuando afecta a un hueso largo, la lesión puede limitarse a la metáfisis o bien afectar a una porción variable de la diáfisis. Las imágenes radiológicas reflejan, de forma significativa, la evolución temporal de las lesiones y la aparición de alteraciones añadidas, en la mayoría de los casos, formación de quistes. De este modo, las lesiones observadas en adultos tienden a mostrar un aspecto más heterogéneo y un mayor componente esclerótico3.
En el cráneo las lesiones de DF afectan típicamente a los huesos faciales y de la base craneal, con esclerosis de aspecto "pagetoide", cuya expresión clínica suele ser asimetría y tumefacción faciales con protrusión en las regiones malar, frontal o temporal. La progresión de estas lesiones puede producir deformidades importantes y compresión de nervios craneales; particularmente grave es la afección de los nervios ópticos.
El tejido óseo displásico de los huesos craneofaciales es propenso a hemorragias, herniación a través de orificios craneales y vías vasculares y a desarrollar quistes poshemorrágicos, lo cual origina múltiples complicaciones.
Las lesiones iniciales que afectan a la columna, la pelvis y las costillas pueden no ser detectadas por las radiografías convencionales; sin embargo, se localizan con exactitud mediante gammagrafía ósea, que es la técnica de imagen con mayor sensibilidad para determinar la extensión de la enfermedad, aunque los hallazgos no son específicos5.
La observación de lesiones rápidamente expansivas con destrucción del hueso cortical debe alertar sobre la posibilidad de degeneración sarcomatosa9.
Las características radiográficas de la DF pueden ser similares a los hallazgos de otras enfermedades óseas que deben considerarse en el diagnóstico diferencial, entre las cuales se encuentran la enfermedad de Ollier, los fibromas no osificantes múltiples, la angiomatosis esquelética y el hiperparatiroidismo primario. Incluso se han descrito cuadros de DF poliostótica que pueden confundirse con metástasis óseas múltiples10. La caracterización cuidadosa del aspecto radiológico de las lesiones, el estudio genético y los hallazgos histopatológicos son los elementos que permiten un diagnóstico correcto en estos casos.
Posibilidades terapéuticasUna vez establecido el diagnostico de DF, las intervenciones traumatológicas y ortopédicas ocupan un papel central en el manejo de estos pacientes y están dirigidas principalmente a consolidación de fracturas, corrección de deformidades óseas y descompresión de los nervios craneales afectados. La afectación del fémur proximal con alto riesgo de fractura se trata de manera más eficaz mediante la inserción de clavos intramedulares, en un intento de prevenir deformidades graves y asimetría en la longitud de las extremidades11. Las intervenciones quirúrgicas craneofaciales sólo estarían indicadas en casos de compresión de nervios craneales con pérdida visual o auditiva12.
Actualmente son limitados los recursos del tratamiento médico de la DF y la literatura aporta datos fundamentalmente con el uso de bisfosfonatos (pamidronato, alendronato y zoledronato)13,14. Se han publicado estudios no controlados en los cuales el tratamiento con pamidronato no obtuvo resultados satisfactorios, mientras que en otros conllevó mejoría del dolor óseo, disminución de marcadores bioquímicos de remodelado óseo y estabilización del aspecto radiográfico de las lesiones15. Así, Plotkin et al16 comunicaron los resultados de la administración de pamidronato intravenoso (1-1,5 mg/kg/día en 3 días consecutivos) en ciclos repetidos cada 4 meses en 18 niños y adolescentes con DF poliostótica. Tras un seguimiento medio de 3,8 años, se observó una disminución de fosfatasa alcalina y NTx; sin embargo, no hubo evidencias radiográficas de relleno de las lesiones líticas ni engrosamiento de la cortical en ningún caso. El estudio histomorfométrico del hueso displásico no mostró diferencias entre quienes recibieron pamidronato (n = 7) y aquellos sin tratamiento (n = 9). Los datos publicados por Chapurlat et al17, de una serie de 58 pacientes con DF (41 adultos y 17 menores de 18 años) tratados con pamidronato 180 mg cada 6 meses, con un seguimiento medio de 50 meses, evidenciaron una significativa disminución del dolor óseo y de los marcadores de remodelado óseo con un incremento de la densidad mineral ósea femoral (en 12 pacientes con afectación de fémur). En el 50% de los casos se observaron signos radiológicos de mejora de las lesiones óseas, con relleno de las lesiones quísticas y/o engrosamiento cortical. No se identificaron factores predictores de respuesta al tratamiento y el perfil de seguridad fue adecuado, documentado especialmente en un subgrupo de pacientes con un seguimiento de más de 8 años. Asimismo, el grupo de estudio de la Sociedad Italiana de Endocrinología Pediátrica comunicó los resultados de una serie de 14 niños y adolescentes con DF moderada o intensa en relación con el sindrome de McCune-Albright que fueron tratados con pamidronato en ciclos cada 4 a 12 meses, según las concentraciones de fosfatasa alcalina. Tras un seguimiento variable (1,9-9 años) se evidenciaron efectos beneficiosos en el dolor óseo y el aspecto radiológico de las lesiones, con una disminución de la incidencia de nuevas fracturas, comparado con el periodo previo al tratamiento18.
En una revisión de los estudios publicados (abiertos y no controlados), Chapurlat13 evaluó los resultados terapéuticos del uso de pamidronato, alendronato y zoledronato en pacientes con DF y concluyó que la administración de bisfosfonatos conlleva disminución de la reabsorción ósea, incremento de la densidad mineral ósea, menor dolor óseo y mejora radiológica de las lesiones displásicas en aproximadamente la mitad de los casos. En pacientes sin respuesta satisfactoria a pamidronato, el cambio a zoledronato (4 mg cada 6 meses, intravenoso) no produjo mejoría clínica o radiológica sustancial. La administración de suplementos de calcio y vitamina D en pacientes con deficiencia estaría justificada para limitar los efectos deletéreos del hiperparatiroidismo secundario y la suplementación con fosfatos podría prevenir los defectos de mineralización en pacientes con tubulopatía con pérdida renal de fosfatos; sin embargo, no se dispone de evidencias de la eficacia de tales suplementos en la DF. El autor concluye que los datos disponibles son limitados por los aspectos metodológicos de los estudios, pero indican efectos beneficiosos de la terapia con bisfosfonatos, aunque son necesarios estudios clínicos controlados para valorar la eficacia de estas intervenciones.
