


Histiocytosis is characterized by proliferation of cells from the mononuclear phagocyte system, and may be divided into Langerhans cell histiocytosis (LCH) and non-Langerhans cell histiocytosis (including Erdheim–Chester disease [ECD]). While diabetes insipidus (DI) is the most common hypothalamic–pituitary consequence, anterior pituitary deficiencies are less known. This study analyzed the frequency and progression of pituitary hormone deficiencies and the radiographic findings in 9 patients (7 with LCH and 2 with ECD) with hypothalamic–pituitary (HP) axis. Eighty-nine percent of patients had DI (62% at diagnosis), and 78% had one or more anterior pituitary deficiencies (71% at diagnosis). HP involvement is relatively common in patients diagnosed with histiocytosis and hormone deficiencies may be present at diagnosis or appear gradually during the course of disease. Regular monitoring of these patients is recommended.
Las histiocitosis son cuadros caracterizados por la proliferación de células del sistema mononuclear fagocítico. Incluyen la histiocitosis de células de Langerhans (HCL) y las histiocitosis de células no Langerhans (entre ellas la enfermedad de Chester–Erdheim [ECE]). Aunque la diabetes insípida (DI) es la alteración hipotálamo hipofisaria (HH) más frecuente, están menos estudiados los déficits hipofisarios anteriores. Se analiza la frecuencia y la progresión de los déficits hormonales hipofisarios y los hallazgos radiológicos de 9 pacientes (7 HCL y 2 ECE) con afectación de la región HH. El 89% de los pacientes presentaba DI (62% al diagnóstico) y el 78%, uno o más déficits anteriores (71% al diagnóstico). Dado que la afectación HH es relativamente frecuente en pacientes con diagnóstico de histiocitosis y que los déficits hormonales pueden estar presentes al diagnóstico o aparecer de forma paulatina durante el curso de la enfermedad, es recomendable monitorizar de manera regular a este tipo de pacientes.
Histiocytoses are a heterogeneous group of diseases characterized by the proliferation of cells from the mononuclear phagocyte system.1 In 1987, the Histiocyte Society classified histiocytosis into three types: class I or Langerhans cell histiocytosis (LCH), class II or non-Langerhans cell histiocytosis (including Erdheim–Chester disease [ECD]) and class III or malignant histiocytosis.2 LCH is the most common and may affect subjects of any age, with a special incidence in childhood at a peak age ranging from 1 to 3 years. In adults, mean age at diagnosis is 33 years.3,4 The estimated overall incidence ranges from 1.9 cases per million and year, with a male/female ratio of 2:1.3 Diagnosis of LCH requires verification of the presence of Birbeck granules by electron microscopy and antigen CD1a or langerin (CD 207) on the cell surface.5 The spectrum of clinical manifestations is diverse, and any tissue or organ may be involved. LCH may be localized, when a single organ is affected (unifocal or multifocal), or systemic.6 Lung and bone are the most commonly involved in adults (in 58% and 57% of cases respectively), followed by skin (36%) and the pituitary gland (29%), while liver, spleen lymph nodes, and bone marrow are less frequently affected.7 Infiltration of the hypothalamic–pituitary axis (HPA) has been reported in 5–50% of necropsies of patients with LCH.8,9 Diabetes insipidus (DI) is the most common endocrine manifestation, occurring in 17–25% of patients,7,10 while anterior pituitary deficiency is seen in 5–20%.9,11 Pituitary involvement may cause, in addition to pituitary dysfunction, neuropsychiatric and behavioral disorders and autonomic and metabolic changes.12
On the other hand, ECD mainly affects adults between the fifth and seventh decades of life. Diagnosis is based on the presence of histiocytes with an absence of Birbeck granules, positive CD68, and negative CD1a.13 Clinical presentation of ECD is polymorphic and heterogeneous. Bone involvement consisting of osteosclerotic lesions in the metaphysis of long bones is characteristic of the disease.14
The purpose of this article was to analyze the frequency and progression of pituitary hormone deficiencies in a series of nine patients with LCH and ECD and HPA involvement.
Material and methodsA retrospective, observational study was conducted to analyze the clinical data from nine patients (5 women and 4 men) with a mean age 52 years (range, 21–74) with a histological diagnosis of histiocytosis and HPA involvement (7 with LCH and 2 with ECD). Median follow-up time was 12 years (range, 4–32).
The variables analyzed included sex, age at diagnosis, disease duration, first symptom, form of disease (localized or systemic), type of endocrine and non-endocrine involvement, treatment, and radiographic findings at symptom start and during the course of the disease.
Diagnosis of LCH was based on verification of the presence of Birbeck granules by electron microscopy and antigen CD1a or langerin (CD 207) on the cell surface.5 ECD was diagnosed based on the absence of Birbeck granules and positive CD68 and negative CD1a tests.13
Hypopituitarism was diagnosed based on the finding of one or more pituitary deficiencies in the following laboratory tests: plasma ACTH, growth hormone (GH), cortisol, prolactin, luteinizing hormone (LH), follicle-stimulating hormone (FSH), serum estradiol and testosterone, measured by a chemiluminescence enzyme assay (Immulite 2000, Siemens), the measurement of thyroid-stimulating hormone (TSH) and free T4 in serum by electrochemiluminescence (Modular E170, Roche), and of serum IGF-I using electrochemiluminescence (IDS-iSYS). For the interpretation of IGF-I, age- and sex-adjusted reference values were used. A diagnosis of DI was made based on hypotonic polyuria (urine output>3.5L/day associated with urinary osmolality<100mOsm/kg) with elevated plasma osmolality and polydipsia.
When baseline hormone measurements were not conclusive for diagnosis, pituitary reserve was studied using dynamic tests: the ACTH rapid stimulation test (Synacthen1-24® 250μg), insulin-induced hypoglycemia and/or the water deprivation test.
Hormone measurements were performed at the start of clinical signs and every 1–2 years for axis re-evaluation.
Hypothalamic involvement was defined as the presence of any of the following signs or symptoms: changes in appetite or thirst, body temperature, sleep pattern, behavior, and recent memory, which was subjectively assessed in all patients.
Magnetic resonance imaging (MRI) of HPA was used for radiographic assessment. Both sagittal and coronal T1- and T2-weighted images were obtained after the administration of gadolinium contrast. Pathological findings in the HPA were assessed using the classification system established for central nervous system lesions in histiocytosis.9
Statistical analysis was performed using software SPSS for Windows version 18, SPSS S.A. Chicago, USA. Values of continuous variables are given as mean and median.
ResultsMedian age at diagnosis was 38 years (range, 3–61). The first symptom was polyuria–polydipsia in five patients, bone pain in two, earache in one, and headache in one patient.
Two of the seven patients with LCH had localized involvement, and five systemic diseases (Table 1).
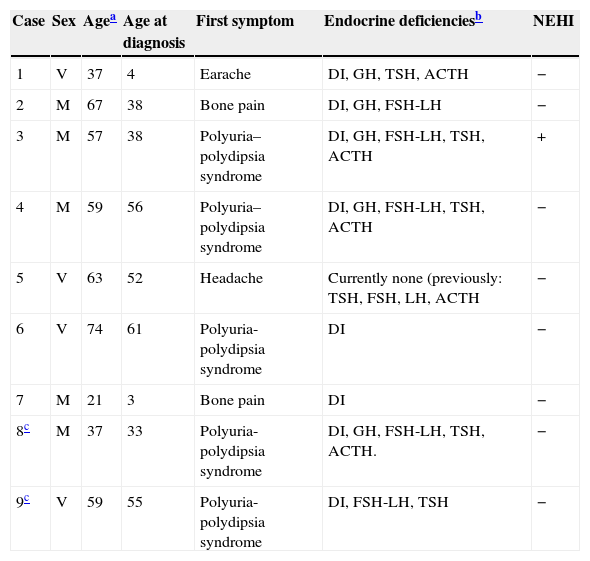
Main clinical characteristics of the nine patients of the series.
| Case | Sex | Agea | Age at diagnosis | First symptom | Endocrine deficienciesb | NEHI |
|---|---|---|---|---|---|---|
| 1 | V | 37 | 4 | Earache | DI, GH, TSH, ACTH | − |
| 2 | M | 67 | 38 | Bone pain | DI, GH, FSH-LH | − |
| 3 | M | 57 | 38 | Polyuria–polydipsia syndrome | DI, GH, FSH-LH, TSH, ACTH | + |
| 4 | M | 59 | 56 | Polyuria–polydipsia syndrome | DI, GH, FSH-LH, TSH, ACTH | − |
| 5 | V | 63 | 52 | Headache | Currently none (previously: TSH, FSH, LH, ACTH | − |
| 6 | V | 74 | 61 | Polyuria-polydipsia syndrome | DI | − |
| 7 | M | 21 | 3 | Bone pain | DI | − |
| 8c | M | 37 | 33 | Polyuria-polydipsia syndrome | DI, GH, FSH-LH, TSH, ACTH. | − |
| 9c | V | 59 | 55 | Polyuria-polydipsia syndrome | DI, FSH-LH, TSH | − |
NEHI: nonendocrine hypothalamic involvement; DI: diabetes insipidus; F: female; M: male.
Two patients had anterior panhypopituitarism at diagnosis, three had one or more anterior deficiencies, and four patients had an intact anterior pituitary gland. After diagnosis, five patients developed one or more anterior deficiencies, with a median of 7.5 years (range, 1–19). The gonadotropin axis was affected in 66.6% of patients, and the somatotropin, corticotropin, and thyrotropin axes in 55.5% of cases. Four patients initially had elevated serum prolactin levels, which were less than four times the upper limit of normal in three, and seven times the upper limit of normal in the remaining patient. One patient recovered all axes five years after the administration of fractionated stereotactic radiation therapy (RT) to the sellar mass. Another patient showed hypothalamic involvement with polyphagia, hypodipsia, hyperthermia, and recent memory impairment. Eight of the nine patients experienced DI. DI was present at diagnosis in five patients, and developed subsequently in the other three patients (two at one year and one at 10 years of diagnosis) (Table 2).
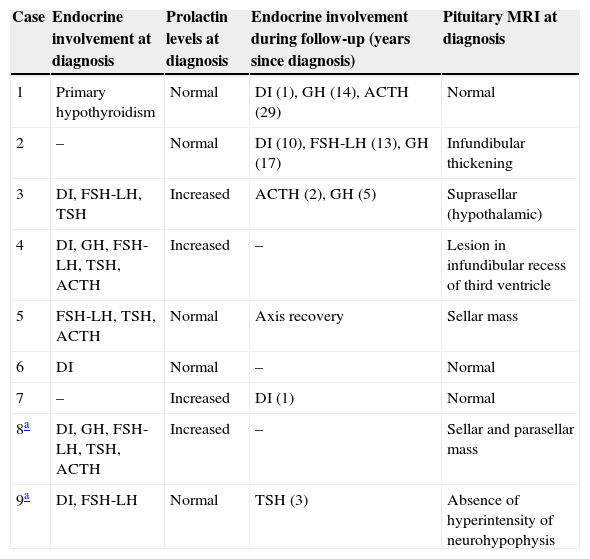
Change over time in hormone deficiencies and pituitary MRI at diagnosis.
| Case | Endocrine involvement at diagnosis | Prolactin levels at diagnosis | Endocrine involvement during follow-up (years since diagnosis) | Pituitary MRI at diagnosis |
|---|---|---|---|---|
| 1 | Primary hypothyroidism | Normal | DI (1), GH (14), ACTH (29) | Normal |
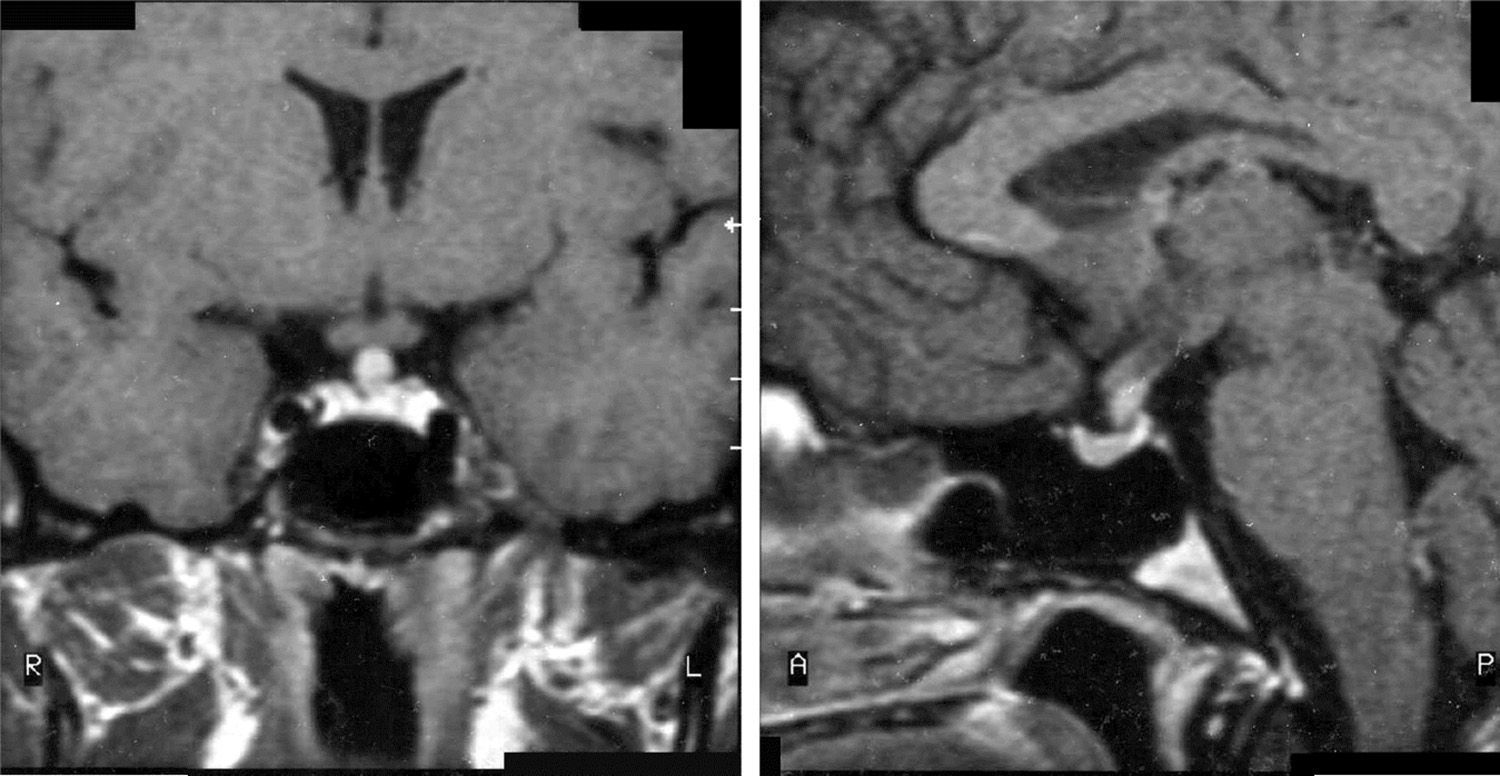
| 2 | – | Normal | DI (10), FSH-LH (13), GH (17) | Infundibular thickening |
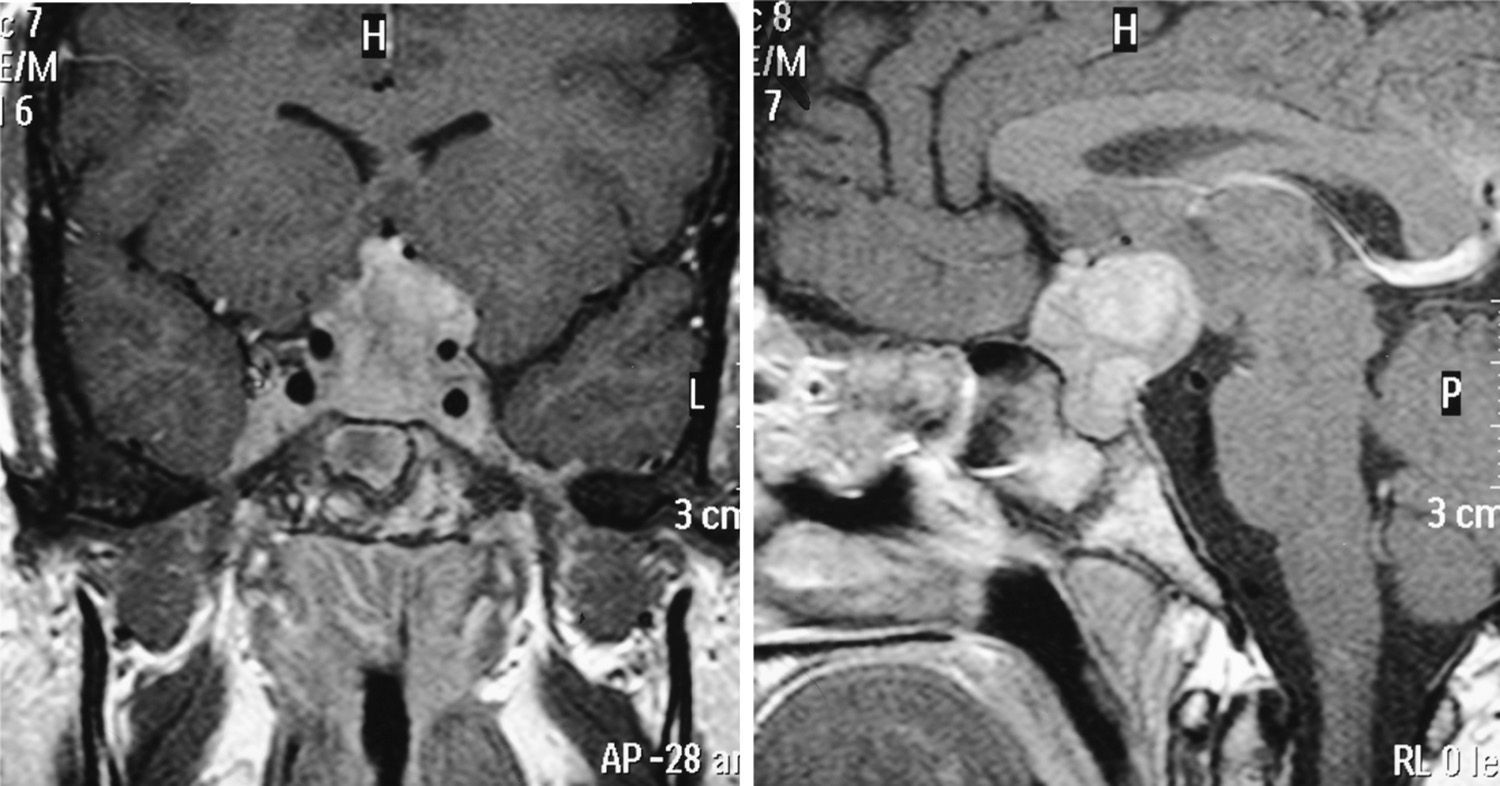
| 3 | DI, FSH-LH, TSH | Increased | ACTH (2), GH (5) | Suprasellar (hypothalamic) |
| 4 | DI, GH, FSH-LH, TSH, ACTH | Increased | – | Lesion in infundibular recess of third ventricle |
| 5 | FSH-LH, TSH, ACTH | Normal | Axis recovery | Sellar mass |
| 6 | DI | Normal | – | Normal |
| 7 | – | Increased | DI (1) | Normal |
| 8a | DI, GH, FSH-LH, TSH, ACTH | Increased | – | Sellar and parasellar mass |
| 9a | DI, FSH-LH | Normal | TSH (3) | Absence of hyperintensity of neurohypophysis |
MRI was pathological at diagnosis in six patients, three with a sellar and/or suprasellar mass, one with pituitary stalk thickening (Fig. 1), one with no hypersignal of neurohypophysis, and one with a lesion of the infundibular recess of the third ventricle. The three patients with normal pituitary MRI at diagnosis showed no radiographic changes during follow-up.

Seven patients had non-endocrine involvement in bone (5 patients), lung (3 patients), orbital soft tissue (2 patients), and skin (1 patient). Multivisceral involvement (bone, orbital tissue, retroperitoneum, mediastinum, and adrenal) was found in one patient.
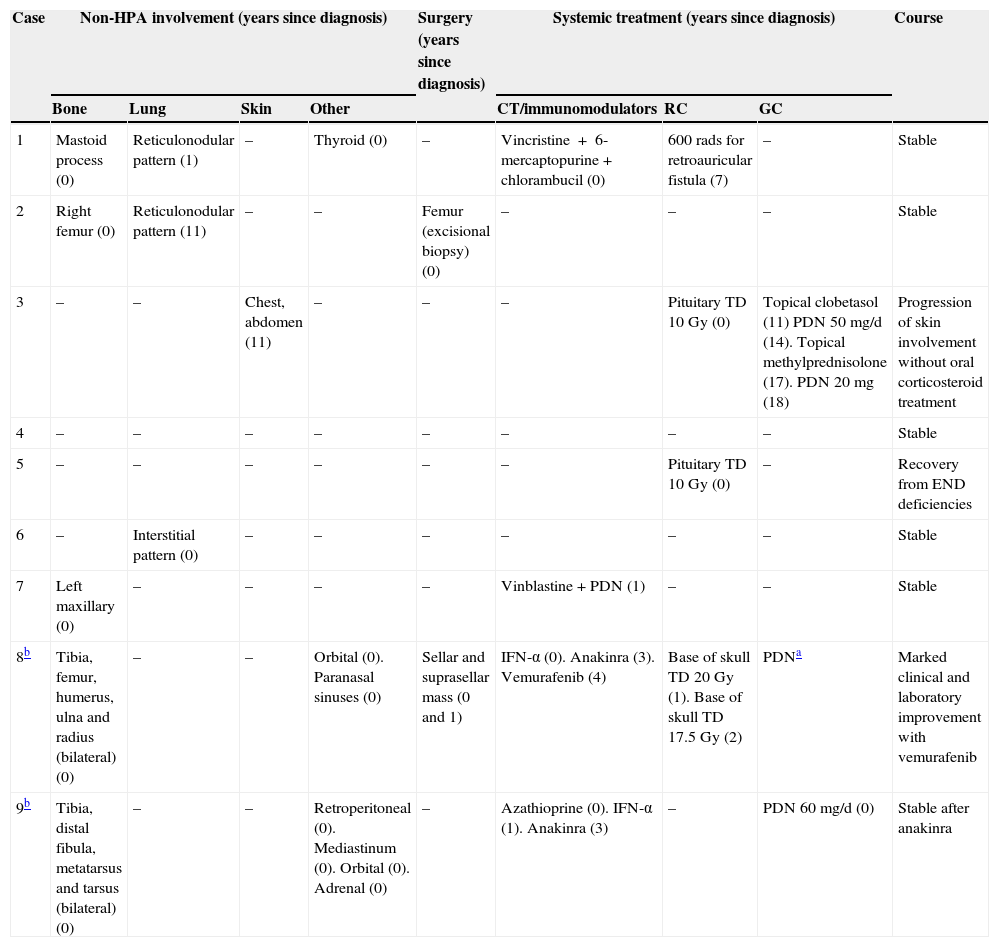
The treatment administered and the course of the disease varied widely depending on organ involvement. Two patients with LCH and sellar and/or suprasellar mass received fractionated stereotactic RT at this level as initial treatment, leading to a stabilization of lesion size. One of them, as already discussed, recovered from all hormone deficiencies five years after treatment. Two patients with LCH with systemic involvement received chemotherapy, which also achieved disease stabilization. A patient with LCH with skin involvement received topical corticosteroid treatment. Three years later, his skin lesions worsened and required oral prednisolone for three years. After the discontinuation of systemic corticosteroid treatment, the skin lesions relapsed despite topical treatment. Treatment with oral prednisone was restarted, leading to the stabilization of the condition, and continues to date. No patient died during follow-up. Table 3 shows the systemic involvement and treatment administered in our series.
Morbidity, treatment, and course.
| Case | Non-HPA involvement (years since diagnosis) | Surgery (years since diagnosis) | Systemic treatment (years since diagnosis) | Course | |||||
|---|---|---|---|---|---|---|---|---|---|
| Bone | Lung | Skin | Other | CT/immunomodulators | RC | GC | |||
| 1 | Mastoid process (0) | Reticulonodular pattern (1) | – | Thyroid (0) | – | Vincristine+6-mercaptopurine+chlorambucil (0) | 600rads for retroauricular fistula (7) | – | Stable |
| 2 | Right femur (0) | Reticulonodular pattern (11) | – | – | Femur (excisional biopsy) (0) | – | – | – | Stable |
| 3 | – | – | Chest, abdomen (11) | – | – | – | Pituitary TD 10Gy (0) | Topical clobetasol (11) PDN 50mg/d (14). Topical methylprednisolone (17). PDN 20mg (18) | Progression of skin involvement without oral corticosteroid treatment |
| 4 | – | – | – | – | – | – | – | – | Stable |
| 5 | – | – | – | – | – | – | Pituitary TD 10Gy (0) | – | Recovery from END deficiencies |
| 6 | – | Interstitial pattern (0) | – | – | – | – | – | – | Stable |
| 7 | Left maxillary (0) | – | – | – | – | Vinblastine+PDN (1) | – | – | Stable |
| 8b | Tibia, femur, humerus, ulna and radius (bilateral) (0) | – | – | Orbital (0). Paranasal sinuses (0) | Sellar and suprasellar mass (0 and 1) | IFN-α (0). Anakinra (3). Vemurafenib (4) | Base of skull TD 20Gy (1). Base of skull TD 17.5Gy (2) | PDNa | Marked clinical and laboratory improvement with vemurafenib |
| 9b | Tibia, distal fibula, metatarsus and tarsus (bilateral) (0) | – | – | Retroperitoneal (0). Mediastinum (0). Orbital (0). Adrenal (0) | – | Azathioprine (0). IFN-α (1). Anakinra (3) | – | PDN 60mg/d (0) | Stable after anakinra |
TD: total dose; END: endocrine; GC: glucocorticoids; HP: hypothalamic–pituitary; IFN-α: interferon alpha; PDN: prednisone; CT: chemotherapy; RT: radiation therapy.
The two patients with ECD were treated with systemic corticosteroids and immunomodulators as described below. Because of the lower frequency of ECD, these two cases in our series are described.
Case 8This was a woman who underwent work-up at another center for left eye proptosis and diplopia in 2003, at the age of 27 years. Laboratory tests showed subclinical hyperthyroidism. Thyroid ophthalmopathy was suspected, and steroid treatment was administered for two years, achieving a remission of the clinical signs. In 2009, the patient attended our hospital complaining of bone pain in the lower limbs over the previous three years and clinical symptoms of polyuria and polydipsia for 10 years. Hormone tests demonstrated DI and anterior panhypopituitarism with hyperprolactinemia. Replacement therapy was therefore started. Pituitary MRI revealed a sellar mass with suprasellar extension (Fig. 2). Subtotal resection was performed, and the histological examination was consistent with ECD. As regards bone involvement, lesions were found in the proximal and distal thirds of the tibia and distal femur in both legs, and the intertrochanteric region of the right femur and humerus, ulna, and radius in both arms. Histological examination of a bone biopsy from the left lower limb was consistent with the same condition. Treatment was started with prednisone and interferon alpha (IFN-α). During follow-up, the sellar and parasellar masses increased in size, requiring repeat surgery and the subsequent administration of fractionated stereotactic RT. Treatment with IFN-α was discontinued and prednisone was continued, with dose tapering. A control pituitary MRI showed an enlargement of the sellar and parasellar masses and the occurrence of tentorial lesions, which required the administration of RT again and the reintroduction of steroid treatment. Treatment was started with anakinra, which was continued for one year with no objective response. After confirmation of the BRAFV600E mutation, the patient started treatment with vemurafenib, which has resulted in marked clinical and laboratory improvement.
Case 9
This was a 59-year-old male patient who reported polyuria-polydipsia syndrome in 2000. Hormonal and biochemical tests showed DI and secondary hypogonadism, for which replacement therapy was started. Pituitary MRI only showed an absence of hypersignal of the neurohypophysis. Bilateral exophthalmos occurred in 2009, and computed tomography (CT) of the orbit showed a retro-orbital mass. Histological examination of a biopsy from the lesion was consistent with ECD. In the extension study, a CT scan of the chest and abdomen revealed retroperitoneal, mediastinal, and adrenal involvement, and a bone scan showed the bilateral involvement of tibia, distal fibula, metatarsus, and tarsus. Treatment was started with glucocorticoids and azathioprine, with no response. IFN-α was therefore added, leading to a partial radiographic response. In 2012, IFN-α was replaced by anakinra, which achieved stabilization of the disease. Thyrotropin deficiency was diagnosed in that same year. Control pituitary MRIs showed no changes.
DiscussionHistiocytosis is a rare disease of unknown etiology with a very heterogeneous clinical presentation. Diagnosis is not made in a substantial proportion of cases due to the variety of nonspecific symptoms. Histiocytosis follows an unpredictable course, sometimes consisting of bouts of the disease. The condition may resolve spontaneously or progress to disseminated disease, compromising vital functions with severe and even fatal consequences.
In adults, as noted above, the most common non-endocrine signs occur in bone, lung, and skin.7 In our series, bone involvement occurred in a proportion of patients similar to that reported in the literature, while lung and skin involvement was less common than in other series.
As regards endocrine manifestations, HPA involvement has been reported in 29.6% of cases in the literature.7 All patients from our series had HPA involvement, as this was a sample selection criterion. Pituitary deficiencies in our study were not exactly comparable to those reported in the literature, which includes patients diagnosed with histiocytosis both with and without HPA involvement. To our knowledge, few studies have selected only patients with a diagnosis of histiocytosis without endocrine involvement.15,16
DI is the most common endocrine deficiency.7,10 When only patients with systemic disease are included, the prevalence of DI may be greater than 40%,14 reaching 94% when another associated hormone deficiency exists. Although DI usually develops within one year of histiocytosis being diagnosed, it may precede such diagnosis, or may occur during the course of the disease. DI is usually permanent.7 In our analysis, DI was found in 89% of patients, and was already present at diagnosis in a little over half of the patients (55%).
Anterior pituitary deficiency has been reported in 20% of patients with LCH, and is almost always associated with DI. Only a few cases of anterior pituitary deficiency without DI have been reported.10 In our series, 78% of patients had some anterior deficiency, which was associated with DI in all but one patient. In this last case, moreover, the deficiencies were reversed after radiation therapy.
GH deficiency is the anterior pituitary deficiency most commonly reported in the literature, affecting more than 42% of patients with LCH and DI.17 It is the first deficiency added in the year subsequent to diagnosis.10 In our analysis, it was the second most common deficiency after gonadotropin deficiency. It was already present at diagnosis in 22% of patients, and occurred during follow-up in all other patients (at 5, 14, and 17 years).
Gonadotropin deficiency is the second most common deficiency reported in the literature.10 Most studies have been conducted on prepubertal patients, and only a few have assessed gonadal function in adults, showing a prevalence ranging from 53% to 58% and always associated with DI.10 Mean latency times are 7 and 9 years after the diagnosis of DI and LCH respectively.10 It was the most common anterior deficiency in our series (67%), and was already present at diagnosis in 83%.
As regards thyrotropin deficiency, in some series it is always associated with panhypopituitarism,10 while in other series it is the third pituitary deficiency in 3.9%, after DI and GH deficiency.18 In the reported series, 55.5% of patients had thyrotropin deficiency, which was associated with panhypopituitarism in 60%, with gonadotropin deficiency and DI in 20%, and with gonadotropin and corticotropin deficiency in 20%.
Corticotropin deficiency has been reported in only 1–2% of patients diagnosed with LCH,17 in most cases in the setting of panhypopituitarism.10 Such a low prevalence may be due to inadequate assessment of this axis and to the fact that cases with partial deficiency have probably been overlooked.16 In our series, corticotropin deficiency occurred with the same frequency as somatotropin and thyrotropin deficiencies (55.5%), and was associated with anterior panhypopituitarism in 60% of cases.
Few studies have measured prolactin secretion in patients with histiocytosis. Moderately elevated serum prolactin levels have been reported in adults with LCH,10 being attributed to infundibular infiltration unrelated to gonadotropin deficiency. Four patients from our series had elevated serum prolactin levels, which were attributed in three of them to stalk compression or infiltration by a sellar and/or parasellar mass. The remaining patient had slightly elevated serum prolactin levels but normal pituitary MRI.
In patients with histiocytosis, the most common hypothalamic change is increased appetite with hyperphagia with resultant obesity.10 In our series, a patient with a suprasellar mass had hypothalamic involvement during follow-up.
Other uncommonly reported nonpituitary endocrine manifestations include primary thyroid involvement due to the infiltration of Langerhans cells,19 primary hypoparathyroidism in the setting of diffuse thyroid and parathyroid infiltration,19 ovarian involvement in cases of disseminated disease,20 adrenal gland infiltration, and direct pancreas involvement in the setting of systemic disease.21 Our series included one patient with primary thyroid involvement in the setting of systemic disease and another patient with ECD with bilateral adrenal infiltration with no associated primary corticotropin deficiency.
As regards imaging tests, pituitary MRI after the administration of gadolinium is the procedure of choice for assessing the HPA region.22 No specific lesion pattern exists in patients with LCH and HPA involvement. The most common radiographic finding is the loss of the hyperintense signal of the neurohypophysis, present in almost all patients with DI.10,22 Other common findings include infundibular thickening, reported in 71% of patients at diagnosis of DI,22 and hypothalamic masses in 8–18% of patients with one or more pituitary deficiencies.10 Pituitary thickening has also been reported in 16% of patients, always associated with DI. Less common findings include partly or completely empty sella turcica and infundibular atrophy.10 The absence of radiographic abnormalities in patients with anterior deficiency has been reported in the literature and has been attributed to microlesions that could lead to hypoperfusion and scar formation, to modulation by cytokines from adjacent bone lesions, or to an autoimmune effect.23
The review of reported studies includes guidelines for the diagnosis and treatment of LCH in patients under 18 years of age.24 For adult patients, recommendations from an expert panel have recently been published.5 Patients with some pituitary hormone deficiency require replacement therapy and regular endocrine assessment to detect any new deficiencies. For localized forms in skin or bone structures, watchful waiting is recommended. When bone involvement exists, if the symptoms require active treatment, low-dose RT, intralesional glucocorticoids or excisional biopsy may be considered.5 Patients with systemic LCH may be treated with chemotherapy schemes including cytostatics such as vinca alkaloids (vinblastine) and glucocorticoids.7 Surgery is reserved for patients with expansive or symptomatic lesions. RT may be used as an adjuvant treatment to surgery. Localized forms of LCH usually have a slow course and a good response to treatment,4 while systemic forms involving liver, spleen, bone or lung, young adults, or poor response to chemotherapy are poor prognostic factors.25
As regards ECD, the first guidelines for its diagnosis and clinical management have recently been published.26 Treatment is reserved for patients with symptomatic disease. Treatment with IFN-α and PEG-IFN-α has been shown to be beneficial for survival, and may therefore be considered as first-line treatment. Anti-cytokine drugs such as anakinra (interleukin-1 receptor antagonist), infliximab (tumor necrosis factor alpha antibody), and tocilizumab (interleukin-6 receptor monoclonal antibody) may be used as second-line treatment, but their benefits have been shown in small patient groups. Chemotherapy treatment using a scheme similar to that used in LCH should also be considered as second line treatment. Glucocorticoids may rapidly decrease edema, but are not effective as monotherapy. In recent years, the BRAFV600E mutation has been reported in more than 50% of patients with ECD. Drugs that inhibit BRAF serine–threonine kinase, such as vemurafenib27,28 and imatinib, have shown promising results in small patient series and clinical trials in terms of clinical and radiographic disease improvement, as occurred in one patient from our series. As regards RT, it is used for localized lesions with compression symptoms, but response is usually less marked than in LCH. Surgery is reserved for resectable cranial lesions or for severe orbital lesions.
In conclusion, HPA involvement is relatively common in patients with histiocytosis. DI is the most common involvement at diagnosis. Other anterior pituitary deficiencies may be present at diagnosis or occur during the course of the disease. Patients with histiocytosis with or without HPA involvement should therefore undergo endocrine work-up at baseline and subsequent regular monitoring for pituitary deficiencies.
Conflicts of interestThe authors state that they have no conflicts of interest.
Please cite this article as: Toro Galván S, Planas Vilaseca A, Michalopoulou Alevras T, Torres Díaz A, Suárez Balaguer J, Villabona Artero C. Alteraciones endocrinas en las histiocitosis de la región hipotálamo hipofisaria. Endocrinol Nutr. 2015;62:72–79.
