




Las histiocitosis son cuadros caracterizados por la proliferación de células del sistema mononuclear fagocítico. Incluyen la histiocitosis de células de Langerhans (HCL) y las histiocitosis de células no Langerhans (entre ellas la enfermedad de Chester-Erdheim [ECE]). Aunque la diabetes insípida (DI) es la alteración hipotálamo hipofisaria (HH) más frecuente, están menos estudiados los déficits hipofisarios anteriores. Se analiza la frecuencia y la progresión de los déficits hormonales hipofisarios y los hallazgos radiológicos de 9 pacientes (7 HCL y 2 ECE) con afectación de la región HH. El 89% de los pacientes presentaba DI (62% al diagnóstico) y el 78%, uno o más déficits anteriores (71% al diagnóstico). Dado que la afectación HH es relativamente frecuente en pacientes con diagnóstico de histiocitosis y que los déficits hormonales pueden estar presentes al diagnóstico o aparecer de forma paulatina durante el curso de la enfermedad, es recomendable monitorizar de manera regular a este tipo de pacientes.
Histiocytosis is characterized by proliferation of cells from the mononuclear phagocyte system, and may be divided into Langerhans cell histiocytosis (LCH) and non-Langerhans cell histiocytosis (including Erdheim-Chester disease [ECD]). While diabetes insipidus (DI) is the most common hypothalamic-pituitary consequence, anterior pituitary deficiencies are less known. This study analyzed the frequency and progression of pituitary hormone deficiencies and the radiographic findings in 9 patients (7 with LCH and 2 with ECD) with hypothalamic-pituitary (HP) axis. Eighty-nine percent of patients had DI (62% at diagnosis), and 78% had one or more anterior pituitary deficiencies (71% at diagnosis). HP involvement is relatively common in patients diagnosed with histiocytosis and hormone deficiencies may be present at diagnosis or appear gradually during the course of disease. Regular monitoring of these patients is recommended.
Las histiocitosis son un grupo heterogéneo de entidades caracterizadas por la proliferación de células del sistema mononuclear fagocítico1. En 1987, la Sociedad Internacional del Histiocito (Histiocyte Society) clasificó las histiocitosis en 3 grupos: clase i o histiocitosis de células de Langerhans (HCL), clase ii o histiocitosis de células no Langerhans (entre ellas la enfermedad de Chester-Erdheim [ECE]) y clase iii o histiocitosis malignas2. La HCL, que es la más frecuente, puede afectar a individuos de cualquier edad, incidiendo especialmente en la infancia con un pico de edad entre 1-3 años. En los adultos, la edad media al diagnóstico es de 33 años3,4. La incidencia global estimada oscila entre 1-9 casos por millón/año, con una proporción hombre:mujer de 2:13. El diagnóstico de HCL requiere la demostración de gránulos de Birbeck mediante microscopia electrónica y la presencia de antígeno CD1a o langerina (CD 207) en la superficie de la célula5. El espectro de las manifestaciones clínicas es variado y cualquier tejido u órgano puede estar afectado. La HCL puede ser localizada cuando afecta a un único órgano (unifocal o multifocal) o sistémica6. En los adultos, la afectación más frecuente es la pulmonar (58%) y ósea (57%), seguida de piel (36%) e hipófisis (29%), mientras que la afectación de hígado, bazo, ganglios linfáticos y médula ósea es menos frecuente7. La infiltración del eje hipotálamo hipofisario (HH) se ha descrito en un 5-50% de las necropsias de pacientes con HCL8,9. El 17-25% de los pacientes desarrollan diabetes insípida (DI), que es la manifestación endocrina más frecuente7,10, seguida del déficit hipofisario anterior en un 5-20%9,11. La afectación hipotalámica puede producir, además de disfunción hipofisaria, trastornos neuropsiquiátricos, del comportamiento y alteraciones autonómicas y metabólicas12.
Por otro lado, la ECE afecta principalmente a adultos entre la quinta y la séptima décadas de la vida. El diagnóstico viene determinado por la presencia de histiocitos con ausencia de gránulos de Birbeck, positividad para CD68 y negatividad para CD1a13. Su presentación clínica es polimorfa y heterogénea, siendo característica de la enfermedad la afectación ósea en forma de lesiones osteoesclerosas en las metáfisis de huesos largos14.
El objetivo de este artículo es analizar la frecuencia y progresión de los déficits hormonales hipofisarios en una serie de 9 pacientes con HCL y ECE con afectación de la región HH.
Material y métodosSe llevó a cabo un estudio observacional retrospectivo donde se analizaron los datos clínicos de 9 pacientes (5 mujeres y 4 varones), con diagnóstico histológico de histiocitosis con afectación de la región HH (7 con HCL y 2 con ECE), con una media de edad de 52 años (rango 21-74). La mediana de seguimiento fue de 12 años (rango 4-32).
Se analizaron las siguientes variables: sexo, edad al diagnóstico, tiempo de evolución, primer síntoma, forma de enfermedad (localizada o sistémica), tipo de afectación endocrina y no endocrina, tratamiento y hallazgos radiológicos al inicio de la clínica y durante la evolución de la enfermedad.
Se utilizó como criterio diagnóstico histológico de HCL la presencia de gránulos de Birbeck mediante microscopia electrónica y del antígeno CD1a o langerina (CD 207) en la superficie de la célula5; y de ECE la ausencia de gránulos de Birbeck y la positividad para CD68 y negatividad para CD1a13.
El hipopituitarismo se diagnosticó ante la presencia de uno o más déficits hipofisarios mediante las siguientes determinaciones analíticas: ACTH plasmática, hormona del crecimiento (GH), cortisol, prolactina, hormona luteinizante (LH), hormona foliculoestimulante (FSH), estradiol y testosterona séricas, medidas con enzimoquimioluminiscencia (Inmulite 2000, Siemens), la determinación de hormona estimulante del tiroides (TSH) y T4 libre séricas mediante electroquimioluminiscencia (Modular E170, Roche) e IGF-I sérico con electroquimioluminiscencia (IDS-iSYS). Para la interpretación de IGF-I, se usaron valores de referencia ajustados a edad y sexo. El diagnóstico de DI se estableció por un cuadro de poliuria hipotónica (diuresis de > 3,5 l/día asociada a una osmolalidad urinaria<100 mOsm/kg) con osmolalidad plasmática elevada y polidipsia.
Cuando las determinaciones hormonales basales no fueron concluyentes para el diagnóstico, se estudió la reserva hipofisaria mediante pruebas dinámicas: prueba de estímulo rápido con ACTH (Synacthen1-24® 250μg), hipoglucemia insulínica y/o prueba de privación de agua.
Las determinaciones hormonales se realizaron al momento del inicio de la clínica y cada 1-2 años para reevaluación de los ejes.
La afectación hipotalámica se definió por la presencia de alguno de los siguientes signos o síntomas: alteraciones del apetito o la sed, de la temperatura corporal, del patrón del sueño, del comportamiento y de la memoria reciente, que se evaluó en todos los pacientes de manera subjetiva.
La evaluación radiológica se realizó mediante resonancia magnética (RM) de la región HH. Se obtuvieron imágenes potenciadas en T1 y T2, tanto en plano sagital como coronal, y tras la administración de contraste con gadolinio. Los hallazgos patológicos en el eje HH se valoraron según el sistema de clasificación establecida para lesiones del sistema nervioso central en histiocitosis9.
El análisis estadístico se realizó mediante el programa SPSS/Windows versión 18, SPSS S.A., Chicago, EE. UU. Los valores de las variables continuas se expresaron en forma de media y mediana.
ResultadosLa mediana de edad al diagnóstico fue de 38 años (rango 3-61). El primer síntoma fue poliuria-polidipsia en 5 pacientes, dolor óseo en 2, otalgia en uno y cefalea en un paciente.
De los 7 pacientes con HCL, 2 presentaban afectación localizada y 5 enfermedad sistémica (tabla 1).
Principales características clínicas de los 9 pacientes de esta serie
| Caso | Sexo | Edada | Edad al diagnóstico | Primer síntoma | Déficits endocrinosb | AHNE |
|---|---|---|---|---|---|---|
| 1 | V | 37 | 4 | Otalgia | DI, GH, TSH, ACTH | – |
| 2 | M | 67 | 38 | Dolor óseo | DI, GH, FSH-LH | – |
| 3 | M | 57 | 38 | Síndrome poliuria-polidipsia | DI, GH, FSH-LH, TSH, ACTH | + |
| 4 | M | 59 | 56 | Síndrome poliuria-polidipsia | DI, GH, FSH-LH, TSH, ACTH | – |
| 5 | V | 63 | 52 | Cefalea | Actualmente ninguno (antes: TSH, FSH-LH, ACTH) | – |
| 6 | V | 74 | 61 | Síndrome poliuria-polidipsia | DI | – |
| 7 | M | 21 | 3 | Dolor óseo | DI | – |
| 8c | M | 37 | 33 | Síndrome poliuria-polidipsia | DI, GH, FSH-LH, TSH, ACTH. | – |
| 9c | V | 59 | 55 | Síndrome poliuria-polidipsia | DI, FSH-LH, TSH | – |
AHNE: afectación hipotalámica no endocrina; DI: diabetes insípida; M: mujer; V: varón.
Dos pacientes presentaban panhipopituitarismo anterior al diagnóstico, 3 tenían uno o más déficits anteriores y 4 presentaban integridad de la hipófisis anterior. Tras el diagnóstico, 5 pacientes desarrollaron uno o más déficits anteriores, con una mediana de 7,5 años (rango 1-19). La frecuencia de afectación de los ejes fue: gonadotropo en un 66,6% y somatotropo, corticotropo y tirotropo en un 55,5%. Inicialmente, 4 pacientes presentaron concentraciones séricas elevadas de prolactina; 3 de ellos tenían valores inferiores a 4 veces el límite superior de la normalidad, y en un caso la cifra de prolactina sérica fue mayor, de 7 veces el límite superior de la normalidad. Un paciente recuperó todos los ejes 5 años después de la administración de radioterapia (RDT) estereotáctica fraccionada a nivel de la masa selar. Otro paciente presentó afectación hipotalámica con polifagia, hipodipsia, hipertermia y afectación de la memoria reciente. Del total de 9 pacientes, 8 presentaron DI. En 5 de ellos estaba presente al momento del diagnóstico y 3 la desarrollaron posteriormente (2 al año y uno a los 10 años del diagnóstico) (tabla 2).
Evolución de los déficits hormonales y RM hipofisaria al diagnóstico
| Caso | Afectación endocrina al diagnóstico | Niveles de prolactina al diagnóstico | Afectación endocrina durante el seguimiento (años tras el diagnóstico) | RM hipofisaria al diagnóstico |
|---|---|---|---|---|
| 1 | Hipotiroidismo primario | Normal | DI (1), GH (14), ACTH (29) | Normal |
| 2 | – | Normal | DI (10), FSH-LH (13), GH (17) | Engrosamiento infundibular |
| 3 | DI, FSH-LH, TSH | Elevada | ACTH (2), GH (5) | Masa supraselar (hipotalámica) |
| 4 | DI, GH, FSH-LH, TSH, ACTH | Elevada | – | Lesión en receso infundibular de tercer ventrículo |
| 5 | FSH-LH, TSH, ACTH | Normal | Recuperación de los ejes | Masa selar |
| 6 | DI | Normal | – | Normal |
| 7 | – | Elevada | DI (1) | Normal |
| 8a | DI, GH, FSH-LH, TSH, ACTH | Elevada | – | Masa selar y paraselar |
| 9a | DI, FSH-LH | Normal | TSH (3) | Ausencia de hiperintensidad de neurohipófisis |
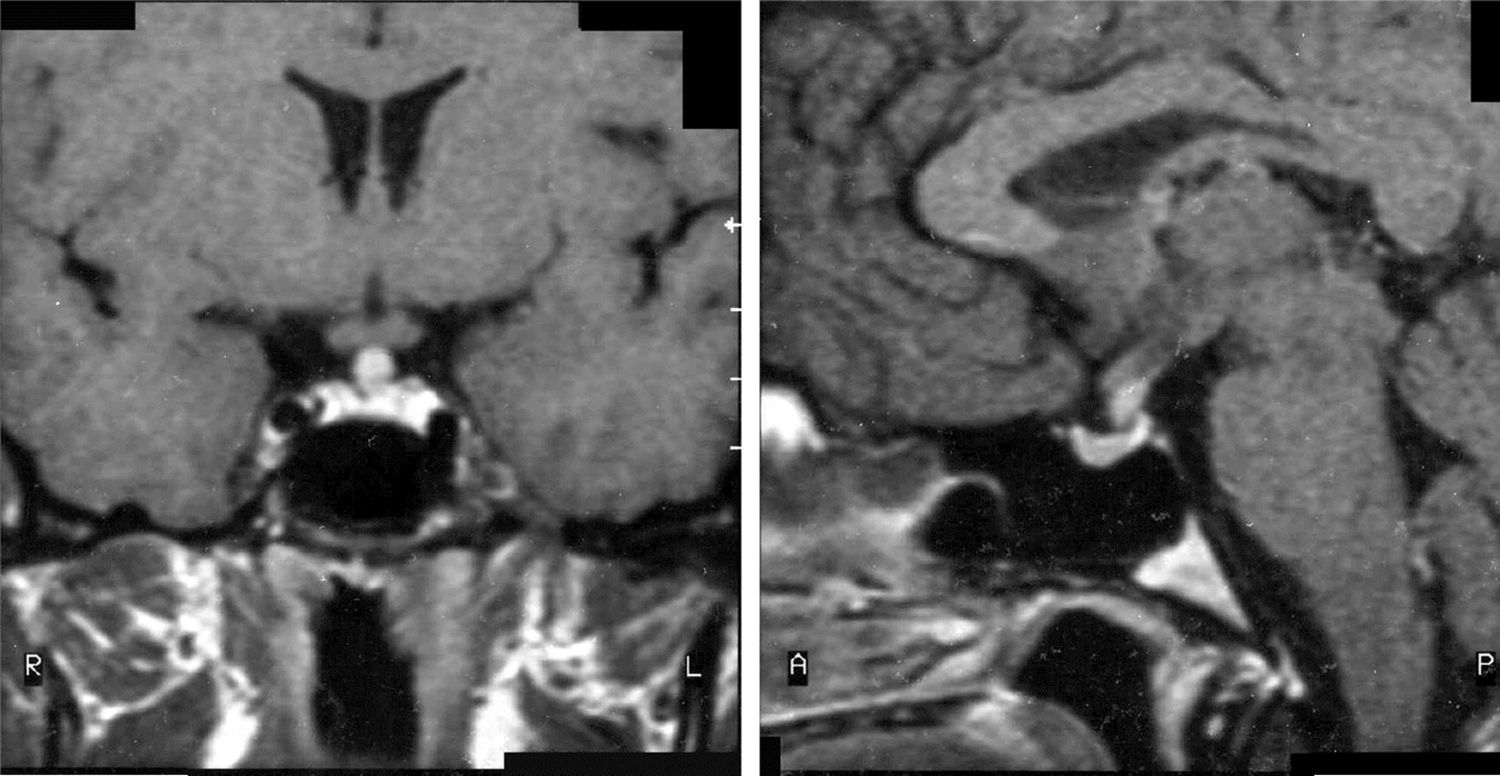
La RM fue patológica al diagnóstico en 6 pacientes: 3 con masa selar y/o supraselar, uno con engrosamiento del tallo hipofisario (fig. 1), uno con ausencia de hiperseñal de la neurohipófisis y uno con lesión del receso infundibular del tercer ventrículo. Los 3 pacientes con RM hipofisaria normal al diagnóstico no presentaron alteración radiológica durante el seguimiento.

Siete pacientes tenían afectación no endocrina: 5 ósea, 3 pulmonar, 2 de tejido blando orbitario y uno cutánea. Un paciente presentó afectación simultánea multivisceral (hueso, tejido orbitario, retroperitoneo, mediastínico y adrenal).
El tratamiento realizado, así como la evolución, fueron muy heterogéneos, dependiendo de la afectación orgánica. Dos pacientes con HCL y masa selar y/o supraselar recibieron RDT estereotáctica fraccionada a este nivel como tratamiento inicial, con estabilización del tamaño lesional. Uno de ellos, tal y como se ha comentado anteriormente, presentó recuperación de todos los déficits hormonales a los 5 años del tratamiento. Dos pacientes con HCL con afectación sistémica recibieron quimioterapia, también con estabilización de la enfermedad. Un paciente con HCL con afectación cutánea recibió inicialmente tratamiento corticoideo tópico. Tres años después, presentó empeoramiento de las lesiones cutáneas, requiriendo la administración de prednisona por vía oral durante 3 años. Tras suspender el tratamiento corticoideo sistémico, a pesar de administrar tratamiento tópico, presentó recidiva cutánea, por lo que se volvió a reintroducir prednisona por vía oral, tratamiento que mantiene en la actualidad con estabilización de la enfermedad. Ningún paciente falleció durante el seguimiento. En la tabla 3 se exponen la afectación sistémica y el tratamiento realizado en nuestra serie.
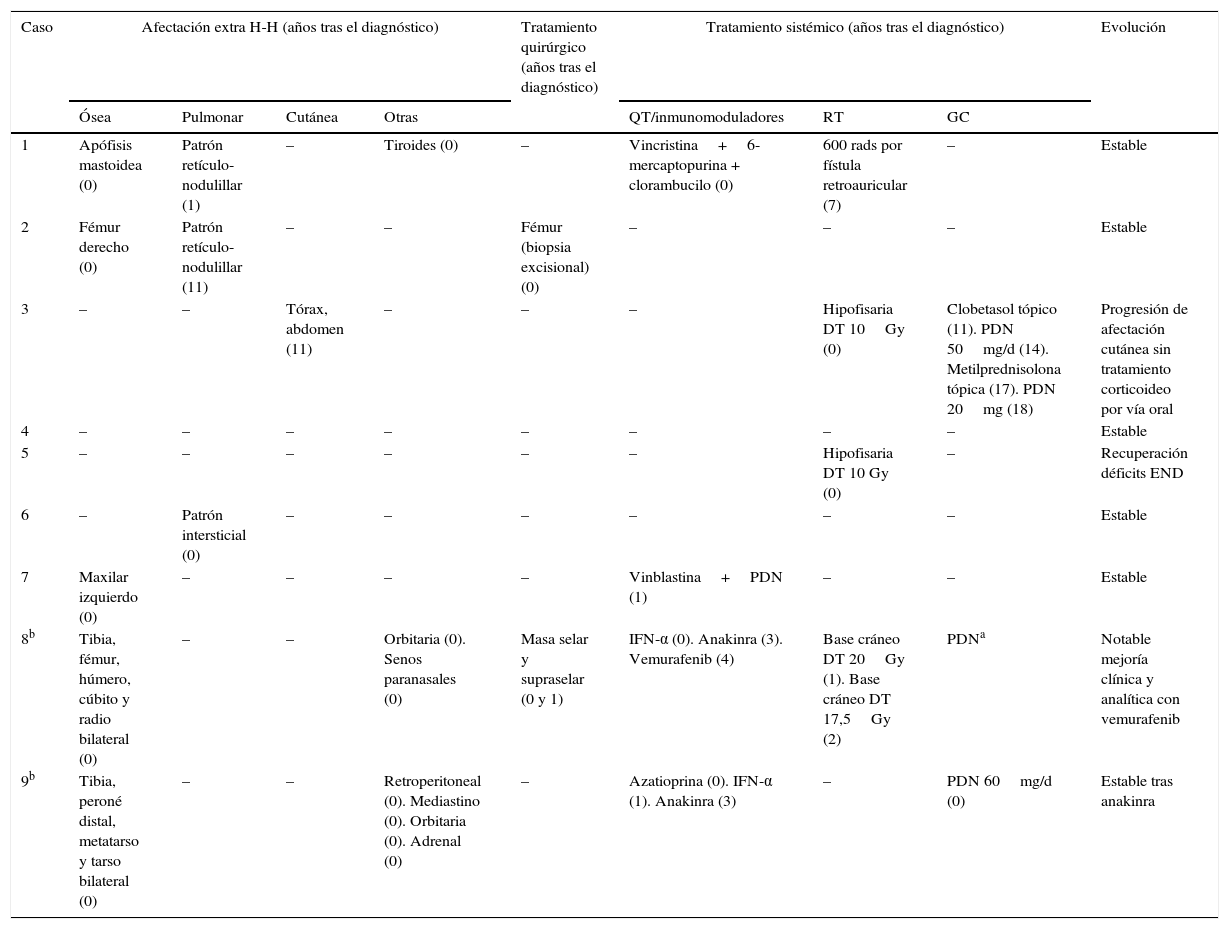
Morbilidad, tratamiento y evolución
| Caso | Afectación extra H-H (años tras el diagnóstico) | Tratamiento quirúrgico (años tras el diagnóstico) | Tratamiento sistémico (años tras el diagnóstico) | Evolución | |||||
|---|---|---|---|---|---|---|---|---|---|
| Ósea | Pulmonar | Cutánea | Otras | QT/inmunomoduladores | RT | GC | |||
| 1 | Apófisis mastoidea (0) | Patrón retículo-nodulillar (1) | – | Tiroides (0) | – | Vincristina+6-mercaptopurina + clorambucilo (0) | 600 rads por fístula retroauricular (7) | – | Estable |
| 2 | Fémur derecho (0) | Patrón retículo-nodulillar (11) | – | – | Fémur (biopsia excisional) (0) | – | – | – | Estable |
| 3 | – | – | Tórax, abdomen (11) | – | – | – | Hipofisaria DT 10Gy (0) | Clobetasol tópico (11). PDN 50mg/d (14). Metilprednisolona tópica (17). PDN 20mg (18) | Progresión de afectación cutánea sin tratamiento corticoideo por vía oral |
| 4 | – | – | – | – | – | – | – | – | Estable |
| 5 | – | – | – | – | – | – | Hipofisaria DT 10 Gy (0) | – | Recuperación déficits END |
| 6 | – | Patrón intersticial (0) | – | – | – | – | – | – | Estable |
| 7 | Maxilar izquierdo (0) | – | – | – | – | Vinblastina+PDN (1) | – | – | Estable |
| 8b | Tibia, fémur, húmero, cúbito y radio bilateral (0) | – | – | Orbitaria (0). Senos paranasales (0) | Masa selar y supraselar (0 y 1) | IFN-α (0). Anakinra (3). Vemurafenib (4) | Base cráneo DT 20Gy (1). Base cráneo DT 17,5Gy (2) | PDNa | Notable mejoría clínica y analítica con vemurafenib |
| 9b | Tibia, peroné distal, metatarso y tarso bilateral (0) | – | – | Retroperitoneal (0). Mediastino (0). Orbitaria (0). Adrenal (0) | – | Azatioprina (0). IFN-α (1). Anakinra (3) | – | PDN 60mg/d (0) | Estable tras anakinra |
DT: dosis total; END: endocrinos; GC: glucocorticoides; H-H: hipotálamo-hipofisaria; IFN-α: interferón alfa; PDN: prednisona; QT: quimioterapia; RT: radioterapia.
Los 2 pacientes con ECE recibieron tratamiento con corticoides sistémicos e inmunomoduladores, tal y como se describe a continuación. Dada la menor frecuencia de ECE dentro de las histiocitosis, se describen los 2 casos de nuestra serie.
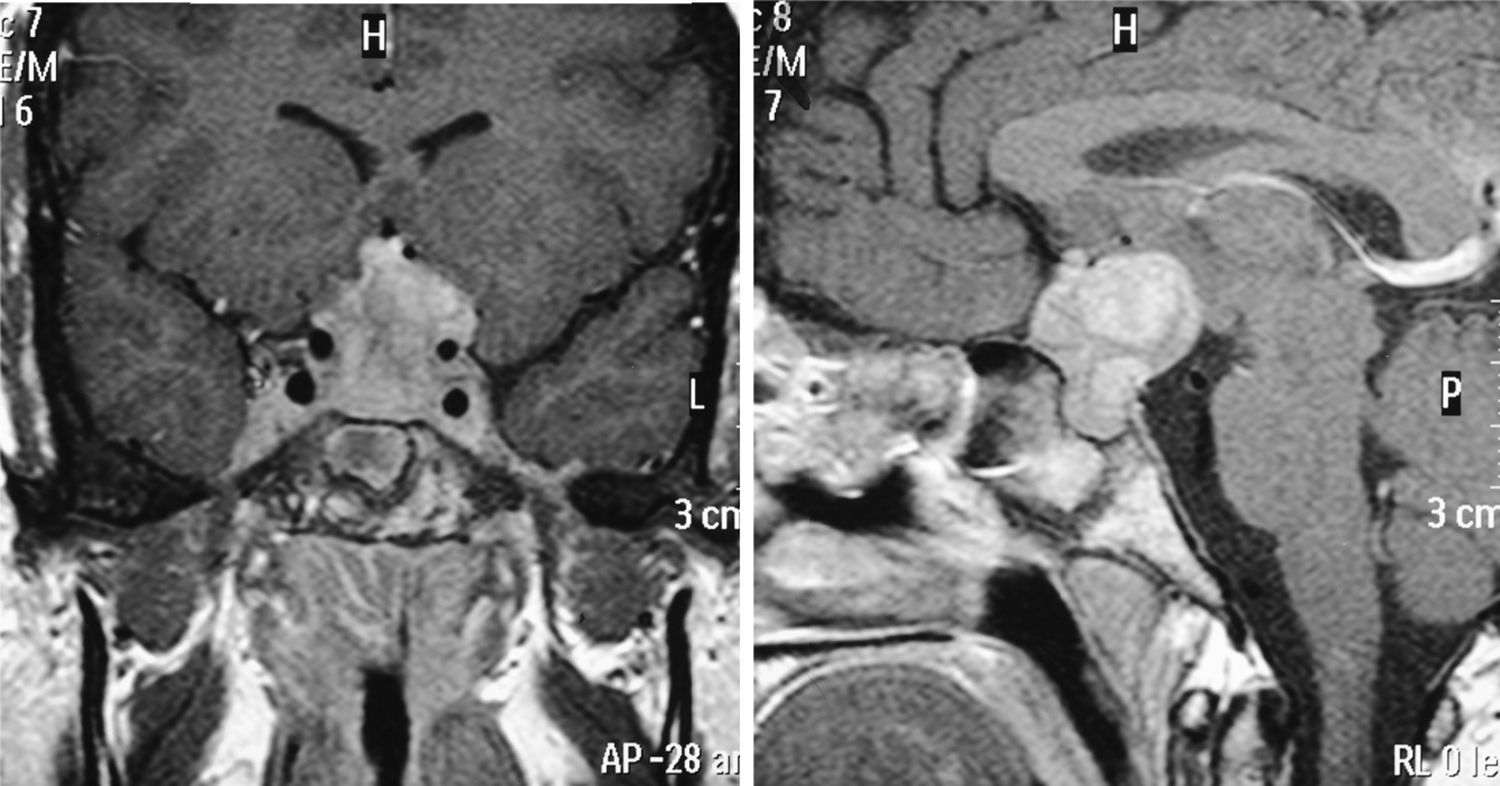
Caso 8Mujer que, en 2003, a la edad de 27 años, fue estudiada en otro centro por proptosis de ojo izquierdo y diplopía. La analítica objetivaba un patrón de hipertiroidismo subclínico, por lo que ante la sospecha de oftalmopatía tiroidea se inició tratamiento esteroideo durante 2 años con remisión de la clínica. En 2009 consultó en nuestro hospital por dolores óseos en las extremidades inferiores de 3 años de evolución y clínica de poliuria y polidipsia desde hacía 10 años. El estudio hormonal puso de manifiesto una DI y un panhipopituitarismo anterior con hiperprolactinemia, por lo que se inició tratamiento sustitutivo de los déficits. La RM hipofisaria objetivó una masa selar con extensión supraselar (fig. 2). Se realizó exéresis subtotal, con diagnóstico histológico compatible con ECE. En cuanto a la afectación ósea, presentaba lesiones a nivel de tercio proximal y distal de tibia y fémur distal bilateral, región intertrocantérea de fémur derecho y húmero, cúbito y radio bilateral. Se realizó biopsia ósea de la extremidad inferior izquierda, con resultado histológico compatible con la misma entidad. Se instauró tratamiento con prednisona e interferón alfa (IFN-α). Durante el seguimiento, presentó crecimiento de la masa selar y supraselar, teniendo que ser reintervenida y administrándose posteriormente RDT estereotáctica fraccionada. Se suspendió el tratamiento con IFN-α y se mantuvo la prednisona, con reducción progresiva de dosis hasta retirarla. La RM hipofisaria de control evidenció un aumento de la masa selar y supraselar, así como aparición de lesiones tentoriales, por lo que fue necesaria de nuevo la administración de RDT y la reintroducción de tratamiento esteroideo. Se inició tratamiento con anakinra, que se mantuvo durante un año sin objetivarse respuesta. Tras haberse confirmado la mutación BRAFV600E, la paciente se halla en tratamiento con vemurafenib, con notable mejoría clínica y analítica.
Caso 9
Varón de 59 años que, en el año 2000, consultó por síndrome de poliuria-polidipsia. El estudio hormonal y bioquímico puso de manifiesto una DI y un hipogonadismo secundario, iniciándose tratamiento sustitutivo de los déficits. La RM hipofisaria mostraba únicamente ausencia de hiperseñal de la neurohipófisis. En 2009 presentó exoftalmos bilateral, por lo que se realizó tomografía computarizada (TC) de órbita que mostraba una masa retroorbitaria. Se realizó biopsia de la lesión, con diagnóstico histológico compatible con ECE. En el estudio de extensión, la TC tóraco-abdominal objetivó una afectación retroperitoneal, mediastínica y adrenal, y la gammagrafía ósea mostró afectación de tibia, peroné distal, metatarso y tarso bilateral. Se instauró tratamiento con glucocorticoides y azatioprina, sin obtener respuesta, por lo que se añadió IFN-α, con discreta respuesta radiológica parcial. En 2012, se sustituyó el IFN-α por anakinra, con estabilización de la enfermedad. Ese mismo año, se diagnosticó de déficit tirotropo. No hubo cambios en las RM hipofisarias de control.
DiscusiónLas histiocitosis son enfermedades raras de etiología desconocida, con una presentación clínica muy heterogénea. En un porcentaje no despreciable de casos no se alcanza el diagnóstico debido a la variedad de síntomas inespecíficos que presentan. Su curso es impredecible y, en ocasiones, evolucionan a brotes; pueden resolverse espontáneamente o progresar a enfermedad diseminada, comprometiendo funciones vitales con graves e incluso fatales consecuencias.
En los adultos, tal y como se ha mencionado anteriormente, la afectación no endocrina más frecuente es a nivel óseo, pulmonar y cutáneo7. En nuestra serie, el porcentaje de afectación ósea es similar a lo descrito en la literatura, a diferencia de la afectación pulmonar y cutánea, que en nuestro caso es menor.
En cuanto a las manifestaciones endocrinas, la afectación del eje HH se ha descrito en la literatura en un 29,6% de los casos7. En nuestra serie, todos los pacientes presentaban dicha afectación, ya que era un criterio de selección de la muestra. En nuestro caso, los déficits hipofisarios no son exactamente comparables con la literatura, ya que lo publicado hace referencia a pacientes con diagnóstico de histiocitosis con y sin afectación HH. Por lo que sabemos, existen pocos estudios que seleccionan únicamente los pacientes con diagnóstico de histiocitosis con afectación endocrina15,16.
La DI es el déficit endocrino más frecuente7,10. Cuando se incluyen solo los pacientes con enfermedad sistémica, la prevalencia de DI puede ser mayor del 40%14, alcanzando el 94% cuando hay asociado otro déficit hormonal. A pesar de que se suele desarrollar en el primer año tras el diagnóstico de histiocitosis, la DI puede preceder a dicho diagnóstico o bien se puede presentar a lo largo del curso de la enfermedad. La DI suele ser permanente7. En nuestro análisis, la DI se objetivó en el 89% de los pacientes y en algo más de la mitad (55%) ya estaba presente al diagnóstico.
El déficit hipofisario anterior se ha descrito en un 20% de los pacientes con HCL y casi siempre está asociado a DI; solo se han evidenciado unos pocos casos de déficit hipofisario anterior sin DI10. En nuestra serie, el 78% de los pacientes presentaba algún déficit anterior y en todos los casos excepto uno estaban asociados a DI; en este último caso, además, los déficits revirtieron tras la radioterapia.
El déficit de GH es el déficit hipofisario anterior más frecuente descrito en la literatura y afecta a más del 42% de los pacientes con HCL y DI17. Es el primer déficit que se añade durante el año posterior al diagnóstico10. En nuestro análisis, es el segundo déficit en frecuencia tras el gonadotropo. En un 22% ya estaba presente al diagnóstico; en el resto, se presentó durante el seguimiento (a los 5, 14 y 17 años).
El déficit gonadotropo es el segundo en frecuencia descrito en la literatura10. La mayoría de los estudios se han realizado en pacientes prepuberales y solo unos pocos han evaluado la función gonadal en adultos, mostrando una prevalencia entre el 53-58% y siempre asociado a DI10. La latencia media es de 7 y 9 años tras el diagnóstico de DI e HCL, respectivamente10. En nuestra serie, es el déficit anterior más frecuente (67%) y en el 83% ya estaba presente al diagnóstico.
Por lo que respecta al déficit tirotropo, en algunas series está siempre asociado a panhipopituitarismo10, mientras que en otras es el tercer déficit hipofisario en un 3,9%, tras la DI y el déficit de GH18. En la presente serie, el 55,5% presentaba dicho déficit; el 60% asociado a panhipopituitarismo, el 20% asociado a déficit gonadotropo y DI, y el 20% asociado a déficit gonadotropo y corticotropo.
El déficit corticotropo se ha descrito en tan solo el 1-2% de los pacientes con diagnóstico de HCL17, la mayoría en contexto de panhipopituitarismo10. El hecho de que la prevalencia sea tan baja puede ser debido a una inadecuada evaluación de este eje y a que, probablemente, han sido obviados casos con déficit parcial16. En nuestra serie, el déficit corticotropo se presenta en igual frecuencia que el somatotropo y tirotropo (55,5%) y en un 60% está asociado a panhipopituitarismo anterior.
Pocos estudios han evaluado la secreción de prolactina en pacientes con histiocitosis. Se han descrito niveles séricos moderadamente elevados en adultos con HCL10 y se atribuye a infiltración infundibular y no relacionada con el déficit gonadotropo. En nuestra serie, 4 pacientes presentan concentraciones séricas elevadas de prolactina, en 3 de ellos atribuibles a compresión o infiltración de tallo por masa selar y/o supraselar y uno con niveles séricos ligeramente elevados pero con RM hipofisaria normal.
En pacientes con histiocitosis, la alteración hipotalámica más frecuente es el aumento del apetito con hiperfagia y con la consecuente obesidad10. En nuestra serie, un paciente con masa supraselar presentó afectación hipotalámica durante el seguimiento.
Otras manifestaciones endocrinas no hipofisarias descritas pero poco frecuentes serían la afectación primaria tiroidea por infiltración de células de Langerhans19, el hipoparatiroidismo primario en contexto de infiltración tiroidea difusa y paratiroidea19, la afectación ovárica en casos de enfermedad diseminada20, la infiltración de glándulas suprarrenales y la afectación directa de páncreas en contexto de enfermedad sistémica21. En nuestra serie, existe un paciente con afectación primaria tiroidea en contexto de enfermedad sistémica y otro paciente con ECE con infiltración adrenal bilateral sin déficit corticotropo primario asociado.
En cuanto a las pruebas de imagen, la RM hipofisaria tras la administración de gadolinio es la prueba de elección para evaluar la región HH22. No existe un patrón específico de lesión en pacientes con HCL y afectación HH. El hallazgo radiológico más frecuente es la pérdida de señal hiperintensa de la neurohipófisis, presente en casi todos los pacientes con DI10,22. Otro hallazgo común es el engrosamiento infundibular, descrito en el 71% de los pacientes en el momento del diagnóstico de DI22 y la presencia de masas hipotalámicas en un 8-18% de los pacientes que presentan uno o más déficits hipofisarios10. El engrosamiento hipofisario también se ha descrito en un 16% de pacientes, siempre asociado a DI. La silla turca parcial o totalmente vacía y la atrofia infundibular son hallazgos menos frecuentes10. La ausencia de anomalías radiológicas en pacientes con déficit anterior se ha descrito en la literatura y se ha atribuido a fenómenos de microlesión que podrían dar lugar a hipoperfusión y formación de cicatrices, a la modulación por parte de citocinas procedente de lesiones óseas adyacentes o bien a un efecto autoinmune23.
En la revisión de los trabajos publicados existe una guía de diagnóstico y tratamiento de la HCL para pacientes menores de 18 años24 y, en el caso de los adultos, se han publicado recientemente unas recomendaciones realizadas por un grupo de expertos5. Los pacientes con algún déficit hormonal hipofisario requieren tratamiento sustitutivo y una evaluación endocrinológica periódica con el fin de detectar la aparición de nuevos déficits. Para las formas localizadas en la piel o las estructuras óseas, se aconseja una conducta expectante. En el caso de la afectación ósea, si los síntomas obligan a un tratamiento activo puede plantearse RDT a dosis bajas, glucocorticoides intralesionales o biopsia excisional5. Los pacientes con HCL sistémica pueden tratarse con esquemas de quimioterapia que incluyen citostáticos como alcaloides de la vinca (vinblastina) y glucocorticoides7. La cirugía se reserva para los pacientes con lesiones expansivas o sintomáticas. La RDT se puede utilizar como tratamiento adyuvante a la cirugía. Las formas localizadas de HCL suelen tener un curso lento y buena respuesta al tratamiento4, mientras que las formas sistémicas con afectación de hígado, bazo, hueso o pulmón, los adultos jóvenes o la escasa respuesta al tratamiento con quimioterapia son factores de mal pronóstico25.
Por lo que respecta a la ECE, se ha publicado recientemente la primera guía de diagnóstico y manejo clínico26. El tratamiento se reserva para aquellos pacientes con enfermedad sintomática. El tratamiento con IFN-α y PEG-IFN-α ha demostrado beneficio en cuanto a supervivencia, por lo que se puede considerar como un tratamiento de primera línea. Los fármacos anticitocina, como anakinra (antagonista del receptor de interleucina-1), infliximab (anticuerpo antifactor de necrosis tumoral alfa) y tocilizumab (anticuerpo monoclonal contra el receptor de interleucina-6) pueden ser utilizados como tratamiento de segunda línea, aunque su beneficio se ha demostrado en grupos reducidos de pacientes. El tratamiento con quimioterapia con esquema similar al utilizado en la HCL se debe considerar también como segunda línea. Los glucocorticoides pueden reducir el edema de manera rápida pero no son efectivos en monoterapia. En los últimos años, se ha descrito la mutación BRAFV600E en más del 50% de los pacientes con ECE, por lo que los fármacos inhibidores de la serina-treonina cinasa BRAF, como vemurafenib27,28 e imatinib, están dando prometedores resultados en series cortas de pacientes y ensayos clínicos en cuanto a mejoría clínica y radiológica de la enfermedad, como sucede con un paciente de nuestra serie. Por lo que respecta al tratamiento con RDT, se utiliza para lesiones focalizadas con clínica compresiva, aunque la respuesta suele ser menor que en la HCL. La cirugía se reserva para lesiones intracraneales resecables o para lesiones orbitarias graves.
En conclusión, la afectación HH es relativamente frecuente en pacientes con histiocitosis. La DI es la afectación más frecuente al diagnóstico. Otros déficits hipofisarios anteriores pueden estar presentes en el momento del diagnóstico o aparecer durante el curso de la enfermedad, por lo que a los pacientes con histiocitosis, con o sin afectación HH, se les debe realizar un estudio endocrinológico inicial y ser monitorizados de manera regular para detectar la aparición de déficits hipofisarios.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
