


El objetivo de nuestro trabajo fue analizar las posibles causas del déficit de hormona de crecimiento (DGH) aislado o en combinación con otras deficiencias hipofisarias, catalogado como idiopático.
Pacientes y métodosEstudiamos a los pacientes con DGH idiopático incluidos en el protocolo de tratamiento con GH sintética para adultos en seguimiento por el Servicio de Endocrinología y Nutrición del Hospital Universitario San Cecilio. Se evaluaron de forma retrospectiva los antecedentes de la historia perinatal, los hallazgos en la resonancia magnética (RM) hipotálamo-hipofisaria y el diagnóstico de DGH y otros déficits hormonales.
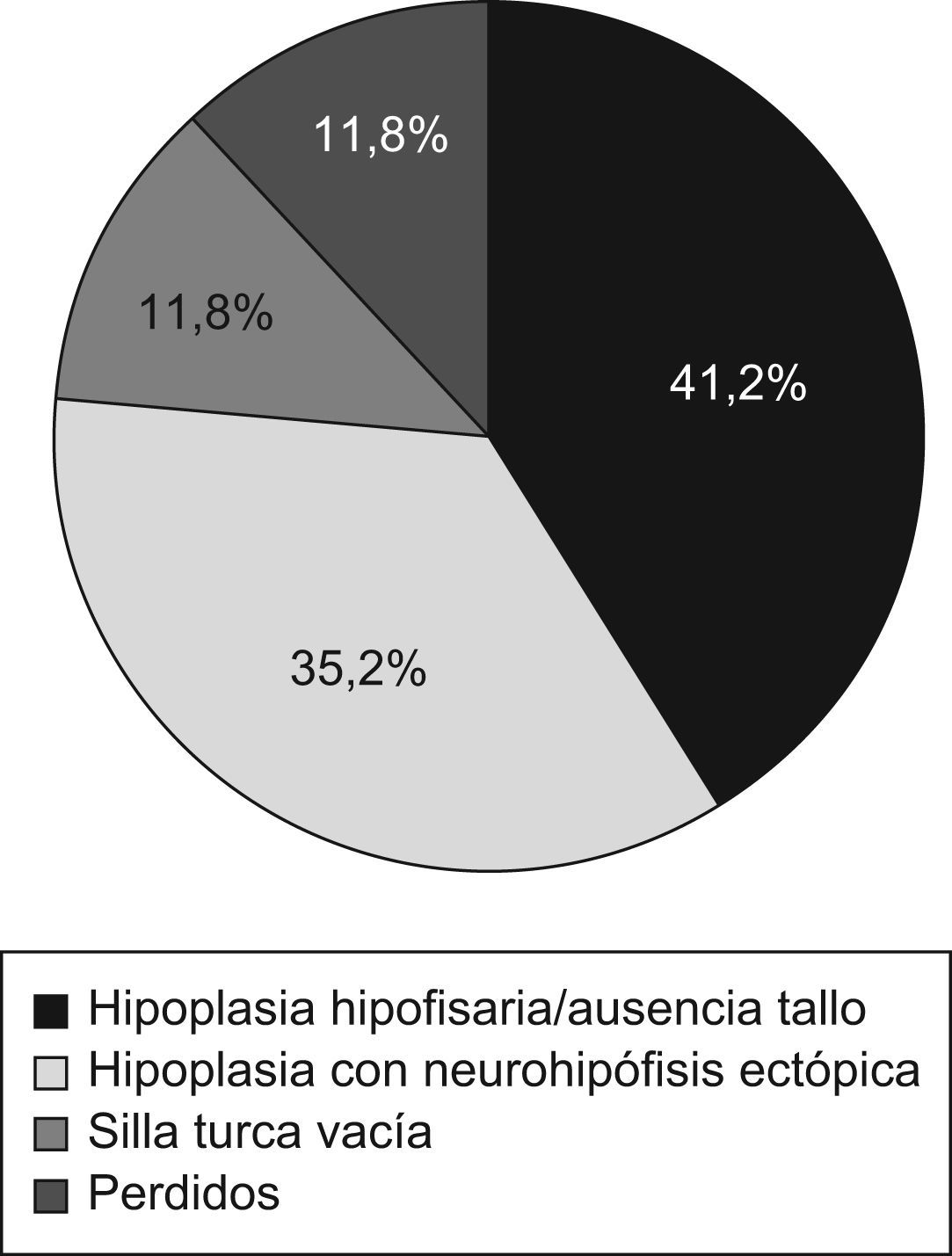
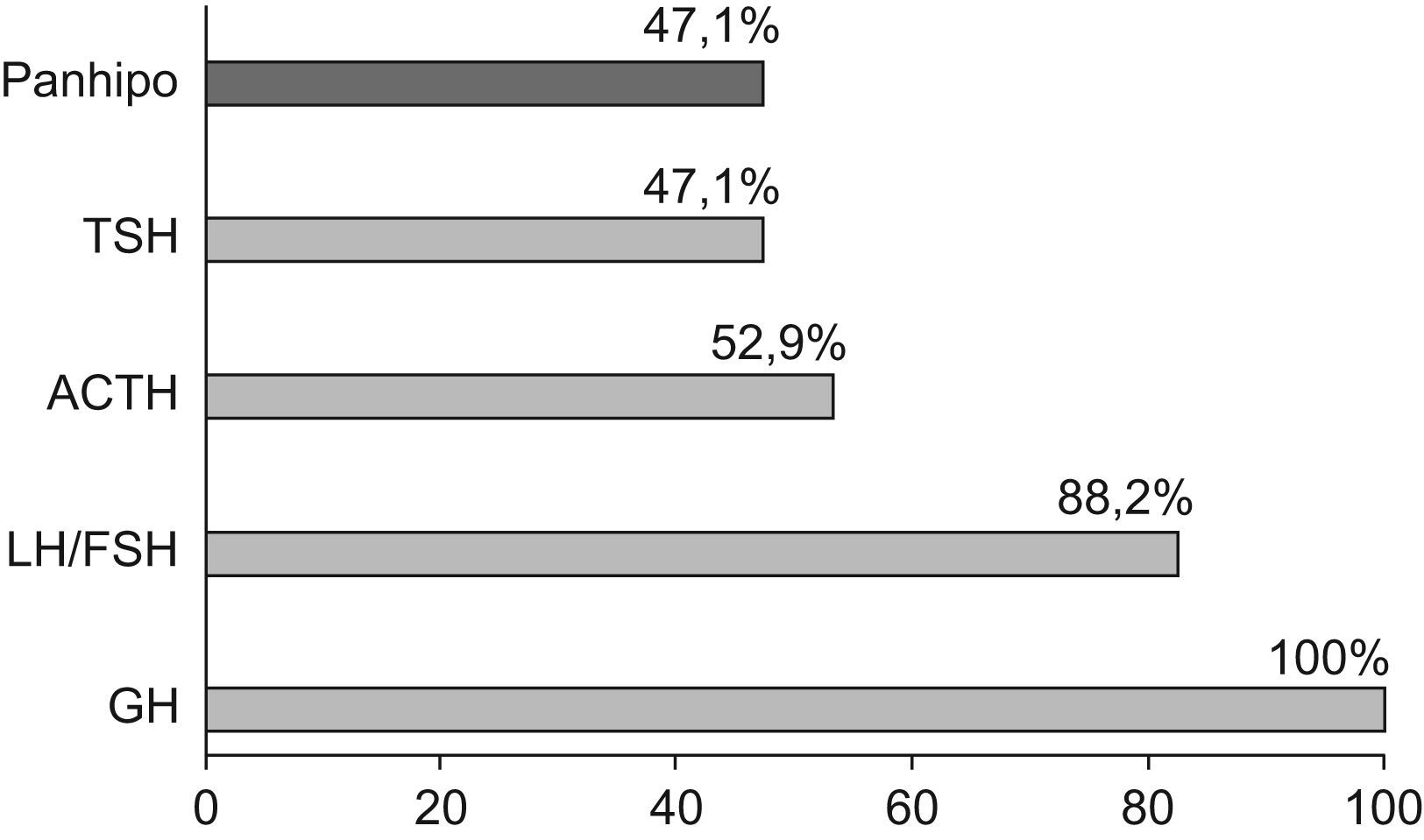
ResultadosSe revisaron las historias de 17 pacientes: 14 varones y 3 mujeres con una edad media al diagnóstico de 8,4±7,3 años. Se presentaron eventos adversos perinatales en 12 pacientes (69,2%). El análisis de las imágenes de RM mostró silla turca vacía (2 pacientes), hipoplasia hipofisaria o ausencia del tallo hipofisario (7 pacientes) e hipoplasia hipofisaria con neurohipófisis ectópica (6 pacientes); en los 2 restantes no se pudo acceder a estos datos. En todos ellos se había establecido el diagnóstico de DGH, en 15 (88,2%) déficit de gonadotropinas, en 9 (52,9%) déficit de corticotropina y en 8 (47,1%) déficit de tirotropina.
ConclusionesEn nuestros pacientes, los eventos adversos durante la gestación o en la historia perinatal y la presencia de alteraciones anatómicas identificadas por RM suponen un marcador de disfunción hipofisaria y pueden ser importantes en la patogenia de esta entidad. El espectro de la enfermedad es variable desde el DGH aislado hasta múltiples déficits hormonales.
To analyze the possible causes of growth hormone (GH) deficiency, whether isolated (GHD) or in combination with other pituitary deficiencies classified as idiopathic.
Patients and methodsWe studied patients with idiopathic GHD included in a protocol of recombinant GH treatment in adults attending the outpatient clinic of the Endocrinology and Nutrition Service of the San Cecilio University Hospital. Perinatal history, findings on magnetic resonance imaging (MRI) of the hypothalamic-pituitary axis and diagnosis of GHD and other deficiencies were retrospectively evaluated.
ResultsA total of 17 patients were included: 14 men and 3 women with a mean age at diagnosis of 8.4±7.3 years. Perinatal adverse events occurred in 12 patients (69.2%). MRI showed empty sella (2 patients), pituitary hypoplasia or absence of the pituitary stalk (7 patients) and pituitary hypoplasia with ectopic posterior pituitary gland (6 patients); in the remaining 2 patients these data were not available. All had an established diagnosis of GHD: 15 with (88.2%) gonadotropin deficiency, 9 (52.9%) with adrenocorticotropic hormone (ACTH) deficiency and 8 (47.1%) with thyroid-stimulating hormone (TSH) deficiency.
ConclusionsIn our patients, adverse events during pregnancy or the perinatal period and the presence of anatomical abnormalities identified by MRI are a marker of pituitary dysfunction and may be important in the pathogenesis of this entity. The clinical spectrum of disease varies from isolated GH deficiency to multiple pituitary hormone deficiencies.
El déficit de hormona de crecimiento (DGH) aislado es una entidad clínica bien conocida que puede asociarse o no con otras deficiencias hormonales hipofisarias. Las causas más frecuentes son los tumores hipofisarios o del sistema nervioso central y el daño cerebral derivado de traumatismos, hemorragias, cirugía o radioterapia1,2. La etiología idiopática supone alrededor del 10–20% según las series, aunque esta proporción está descendiendo al identificarse nuevos factores causales3. De esta forma, la investigación de eventos adversos durante el periodo perinatal, los estudios genéticos actuales y los avances en las técnicas de imagen ayudan en la comprensión de la etiopatogenia de esta enfermedad.
En el desarrollo hipofisario intervienen agentes ambientales y factores genéticos de transcripción. Se han evidenciado relaciones causales entre la presencia de complicaciones durante la gestación y el periodo neonatal y el hipopituitarismo4. Del mismo modo, numerosos estudios sugieren que las mutaciones de los genes encargados de la codificación del eje hipotálamo-hipofisario pueden correlacionarse con las alteraciones neuroanatómicas halladas en estos pacientes5.
La resonancia magnética (RM) es una técnica no invasiva que facilita la visualización de la hipófisis y el tallo, siendo posible relacionar diversas alteraciones neuroanatómicas con las deficiencias hormonales hipofisarias6.
Nuestro estudio supuso un análisis de las posibles causas del DGH aislado o en combinación con otras deficiencias hormonales hipofisarias, catalogado como idiopático, centrándonos en la influencia de los factores ambientales y en las anomalías neuroanatómicas del área hipotálamo-hipofisario.
Pacientes y métodosSe trata de un estudio observacional retrospectivo y descriptivo de 17 pacientes diagnosticados de DGH idiopático e incluidos en un protocolo de tratamiento con GH sintética para adultos en seguimiento por el Servicio de Endocrinología y Nutrición del Hospital Universitario San Cecilio de Granada.
Las variables recogidas de la historia clínica fueron sexo, edad actual y edad al diagnóstico del DGH, peso, talla e índice de masa corporal (calculado por la fórmula de Quetelet) actuales, antecedentes familiares de enfermedad hipofisaria, datos obstétricos con antropometría al nacimiento y edad gestacional, uso de fármacos en el embarazo y presencia de eventos adversos perinatales, estudio clínico y bioquímico de los déficits hormonales hipofisarios y hallazgos en el estudio de imagen de RM hipotálamo-hipofisaria.
Se consideraron como eventos adversos perinatales la prematuridad, la amenaza de aborto durante la gestación, el parto distócico, los signos de sufrimiento fetal con test de Apgar inferior a 8 a los 5min del nacimiento y las infecciones graves con necesidad de ingreso hospitalario durante el primer mes de vida.
Se registraron los estudios hormonales considerando diagnóstico de DGH en la infancia un pico de GH en respuesta a dos pruebas de estímulo farmacológicas (clonidina, insulina, arginina o L-dopa) inferior a 10ng/ml, y en el adulto un pico de GH en respuesta a la prueba de hipoglucemia insulínica inferior a 5ng/ml (deficiencia grave si era <3ng/ml). La valoración del eje tirotropo se llevó a cabo mediante la determinación de las concentraciones séricas de tiroxina no unida a proteínas, triyodotironina no unida a proteínas y tirotropina, y se realizó un test de estimulación con tiroliberina en casos excepcionales. El eje adrenal se evaluó mediante las determinaciones de cortisol sérico y corticotropina plasmática basales (a las 8h), y de la respuesta del cortisol sérico a la hipoglucemia inducida por insulina, y el eje gonadotropo mediante la evaluación clínica del estadio puberal y la determinación sérica de gonadotropinas, estradiol y testosterona.
A todos los pacientes se les realizó una RM hipotálamo-hipofisaria con contraste intravenoso (gadolinio) en planos coronal, sagital y axial en secuencias T1 y T2.
El programa estadístico utilizado fue SPSS versión 15.0. Las variables cuantitativas se expresan como media±desviación estándar y las dicotómicas como porcentaje.
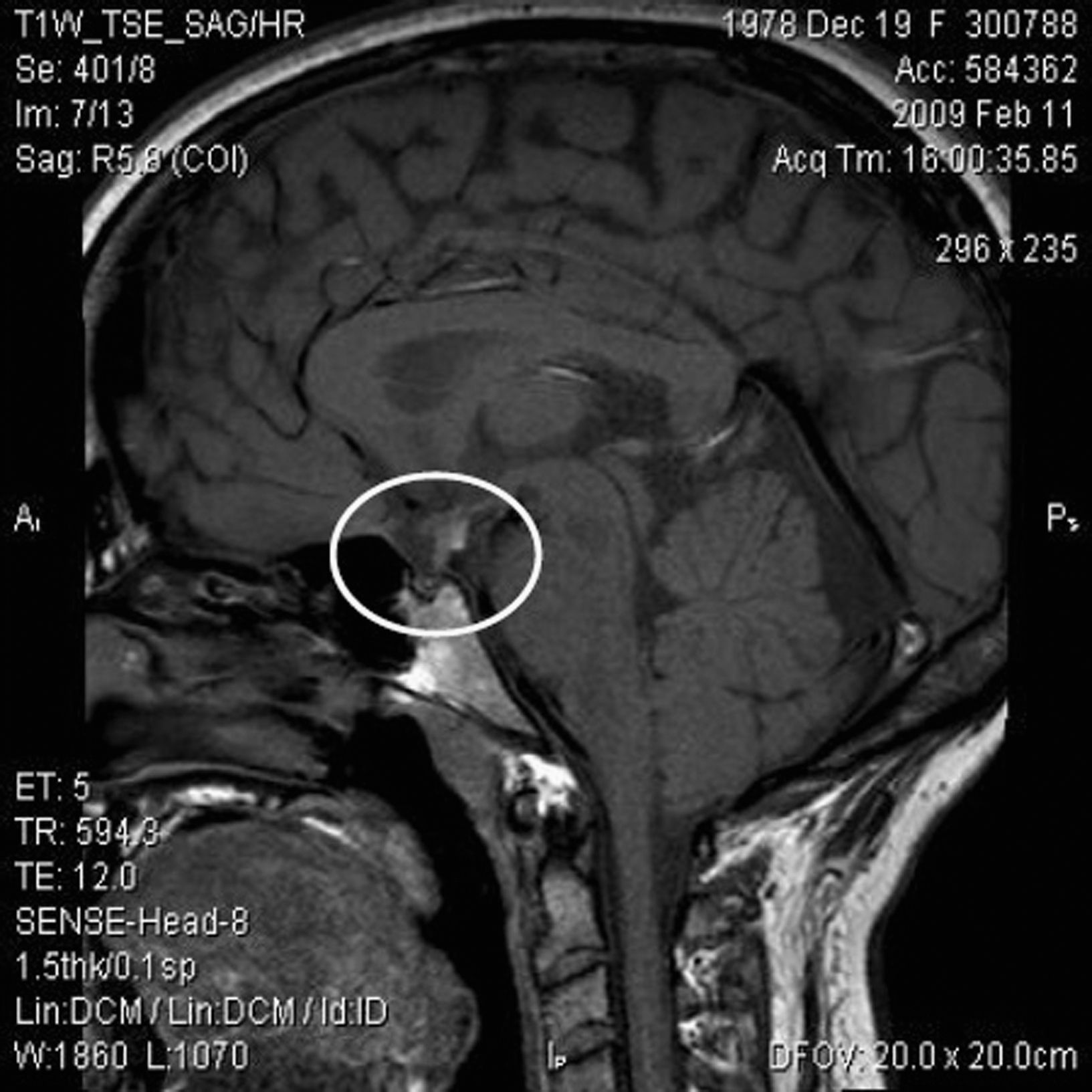
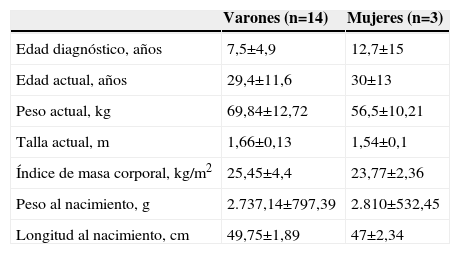
ResultadosSe revisaron las historias clínicas de 17 pacientes (32,1%) de los 53 incluidos en el protocolo de tratamiento con GH sintética para adultos, catalogados de DGH idiopático y en seguimiento por el Servicio de Endocrinología y Nutrición del Hospital Universitario San Cecilio. Se trataba de una muestra de 14 varones y 3 mujeres con una edad media actual de 29,5±11,5 años y una edad media al diagnóstico de 8,4±7,3 años, cuyas características antropométricas actuales y al nacimiento se reflejan en la tabla 1. En ningún caso se constataron antecedentes familiares de enfermedad hipofisaria ni uso de fármacos durante la gestación. El 69,2% (12 pacientes) presentó eventos adversos perinatales. Al analizar las imágenes de RM, se encontró a 2 pacientes con silla turca vacía, a 7 con hipoplasia hipofisaria o ausencia del tallo hipofisario y a 6 con hipoplasia hipofisaria y neurohipófisis ectópica (fig. 1); en los 2 pacientes restantes no se pudo acceder a estos datos (fig. 2). Todos los pacientes presentaban diagnóstico de DGH; el 88,2% (15 pacientes), déficit de gonadotropinas; el 52,9% (9 pacientes), déficit de corticotropina, y el 47,1% (8 pacientes), déficit de tirotropina (fig. 3).
Características antropométricas actuales y al nacimiento
| Varones (n=14) | Mujeres (n=3) | |
| Edad diagnóstico, años | 7,5±4,9 | 12,7±15 |
| Edad actual, años | 29,4±11,6 | 30±13 |
| Peso actual, kg | 69,84±12,72 | 56,5±10,21 |
| Talla actual, m | 1,66±0,13 | 1,54±0,1 |
| Índice de masa corporal, kg/m2 | 25,45±4,4 | 23,77±2,36 |
| Peso al nacimiento, g | 2.737,14±797,39 | 2.810±532,45 |
| Longitud al nacimiento, cm | 49,75±1,89 | 47±2,34 |
Los datos están expresados como media±desviación estándar.



En nuestro trabajo se estudió a pacientes diagnosticados de DGH idiopático y en tratamiento con GH sintética incidiendo en la frecuencia de eventos adversos en su historia perinatal, valorando los defectos neuroanatómicos identificados mediante RM hipotálamo-hipofisaria e identificando la asociación con otros déficits hormonales.
Los factores ambientales contribuyen en la génesis hipofisaria, y en diversos estudios se ha evidenciado una posible relación causal entre las complicaciones gestacionales-perinatales y el hipopituitarismo destacando la amenaza de aborto, el parto de nalgas y otras distocias, la asfixia o el sufrimiento neonatal y las infecciones graves durante el primer mes de vida. En nuestra muestra hay que destacar la alta prevalencia de eventos adversos perinatales, que alcanzó el 69,2%, mayor a la descrita para la población general, entre el 15–30% según la bibliografía4. No obstante, es difícil diferenciar si las alteraciones perinatales y del parto causan disrupciones traumáticas del tallo hipofisario o si los defectos estructurales y funcionales del eje incrementan la prevalencia de las complicaciones perinatales7.
Adicionalmente, se han implicado mutaciones de los genes encargados de la diferenciación y la proliferación celular del eje somatotropo y de la secreción de GH (GH1 y GHRHR)8, y se han considerado fenotipos endocrinos y neurorradiológicos en pacientes con mutaciones de los factores de transcripción hipofisarios PROP1, HESX1, LHX4 y SOX39. Los análisis genéticos pueden jugar un papel novedoso en el entendimiento de los mecanismos patogénicos del hipopituitarismo y la predicción de la evolución de estas alteraciones. En los pacientes de nuestra muestra no se realizó un estudio genético puesto que no está estandarizada su determinación para el diagnóstico, siendo prioritarias la evaluación clínica, bioquímica y neurorradiológica. Algunos autores proponen limitar las indicaciones del estudio genético a pacientes con retraso del crecimiento grave y antecedentes familiares de talla baja. Asimismo, no existe consejo genético establecido en caso de mutaciones específicas8.
Con la ventaja de las imágenes de RM y los avances en el entendimiento del desarrollo hipofisario ha sido posible relacionar las alteraciones neuroanatómicas con las deficiencias hormonales10. Tanto es así, que puede dudarse del diagnóstico de hipopituitarismo permanente si la glándula hipofisaria es normal. Los hallazgos principales en los pacientes de nuestro trabajo fueron la hipoplasia hipofisaria anterior, la ectopia de la neurohipófisis y la ausencia de tallo hipofisario, alteraciones definidas como predictoras de hipopituitarismo7,11.
Tanto la presencia de condiciones desfavorables perinatales como las alteraciones genéticas y las anomalías neuroanatómicas contribuyen a la variabilidad clinicofuncional con una mayor frecuencia de déficits hormonales múltiples12. En nuestro estudio, el DGH aislado solo se presentó en el 11,8% de los pacientes, mientras que existía panhipopituitarismo en el 47,1% y déficit combinado de GH y gonadotropinas en el 88,2%.
En conclusión, los eventos adversos durante la gestación o en la historia perinatal y la presencia de alteraciones anatómicas identificadas por RM suponen un marcador de disfunción hipofisaria y pueden ser importantes en la patogenia del DGH aislado o combinado con otras deficiencias hormonales. El espectro clínico es variable, siendo frecuentes los déficits hormonales hipofisarios múltiples, por lo que es fundamental la reevaluación funcional periódica en los pacientes diagnosticados de DGH.
La realización en el futuro de estudios multicéntricos permitirá analizar un número mayor de pacientes, pudiendo establecer diferencias epidemiológicas, clínicas y diagnósticas en función de las circunstancias desfavorables perinatales y los hallazgos de neuroimagen. Asimismo, con investigaciones adicionales sobre la influencia genética se podrían establecer las indicaciones del análisis de los diversos genes implicados y unas directrices de consejo genético para los pacientes afectos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
