





Los craneofaringiomas son tumores epiteliales raros localmente agresivos que habitualmente se localizan en la región selar y supraselar. El diagnóstico de sospecha del craneofaringioma se basa en los hallazgos clínicos y radiológicos, posteriormente confirmados con el estudio histológico. El tratamiento de elección es la cirugía en la gran mayoría de los casos, con objeto de reducir los síntomas compresivos y resecar la mayor parte de la masa tumoral con la menor morbilidad posible. La radioterapia externa es el tratamiento estándar para el control de restos tumorales poscirugía o recidivas locales.
Las lesiones paraselares son lesiones de prevalencia muy baja y engloban enfermedades neoplásicas, inflamatorias, infecciosas, embrionarias y vasculares. Su diagnóstico y tratamiento es específico según el tipo de lesión
Craniopharyngiomas are rare, locally aggressive epithelial tumors usually located in the sellar and suprasellar region. Diagnosis of craniopharyngioma is usually suggested by clinical and radiological findings that should be confirmed histologically. Surgery is the treatment of choice for most patients. The goal of surgery is to relieve compressive symptoms and to remove as much tumor as safely possible. Radiation therapy is the usual treatment to control postoperative tumor remnants and local recurrences.
Parasellar lesions are low prevalent lesions and include neoplastic, inflammatory, infectious, developmental, and vascular diseases. Both their diagnosis and treatment depend on the type of lesion.
Esta guía se ha elaborado a raíz de la propuesta del Área de Conocimiento de Neuroendocrinología (GNE) de la SEEN de actualización de las Guías de Práctica Clínica en Neuroendocrinología como puesta al día de una guía previa realizada en el año 20071. Cuando ha sido posible se ha evaluado la calidad de la evidencia y el peso de las recomendaciones de acuerdo con el sistema propuesto por la Agency for Health Care Policy and Research2 (tabla 1). No obstante, ante la ausencia de ensayos clínicos o metaanálisis, las recomendaciones de esta guía se basan principalmente en la evaluación de las series de casos más relevantes publicados en las principales revistas médicas internacionales y opiniones de expertos. Esta guía no debe ser considerada como un estándar de práctica clínica, sino como una herramienta para ayudar a los profesionales implicados en el manejo de estos pacientes, en base a la evidencia médica disponible hasta la actualidad (anexo 1).
IntroducciónLos craneofaringiomas son tumores epiteliales raros localmente agresivos que habitualmente se localizan en la región selar y supraselar. Derivan de restos embrionarios del conducto craneofaríngeo formado por la evaginación ectodérmica que constituye la bolsa de Rathke y que origina la adenohipófisis primitiva. Aunque de naturaleza benigna (tipoi de la OMS), resultan localmente invasivos; infiltran los órganos y estructuras adyacentes como el hipotálamo, la hipófisis, el quiasma y los nervios ópticos, y originan, per se o tras su tratamiento, una considerable morbimortalidad. Solo excepcionalmente se ha descrito transformación maligna3,4. Aunque a medio plazo su pronóstico es mejor en pacientes jóvenes que en adultos (supervivencia del 83-96% vs. el 54-96% a 5años), a largo plazo no se han encontrado diferencias sustanciales (62% vs. 66-85% a 20años). No obstante, el progreso de las técnicas diagnósticas y terapéuticas ha mejorado las tasas de supervivencia y reducido las complicaciones5.
EpidemiologíaLos craneofaringiomas son tumores poco frecuentes. Representan el 1-3% de los tumores intracraneales, con una incidencia de 0,5-2 casos por millón de personas/año y una prevalencia estimada de 1-3 por 100.000 habitantes. Presentan una distribución bimodal, con un pico de incidencia entre los 5 y los 15años y otro en la quinta década de la vida. Son la neoplasia no neuroepitelial intracraneal más frecuente en niños (entre el 5,3 y el 15%)6-9 y la neoplasia de la fosa hipofisaria más frecuente a esta edad (80-90%). Más del 50% de los casos de su variante principal (adamantinomatosa) se diagnostican en pacientes menores de 20años. Los tumores confinados exclusivamente a la región intraselar constituyen el 5-6% de los casos, mientras que el 94-95% presentan extensión supraselar (puramente supraselar el 20-41%, y paraselar, afectando a la fosa anterior, la fosa media, el clivus y la fosa posterior entre el 20 y el 30% de los casos)3,8-10.
Anatomía patológicaLos craneofaringiomas son tumores sólidos con un componente quístico variable, estrechamente adheridos a las estructuras vasculares y el tejido cerebral adyacente a expensas de nidos celulares infiltrados en la zona de gliosis reactiva peritumoral.
Se han descrito 2 tipos histológicos fundamentales y una forma mixta o transicional.
La forma adamantinomatosa es la más frecuente (85%). Deriva de restos epiteliales de tipo ameloblastoma de la cavidad orofaríngea primitiva y predomina en niños (95%). Se caracteriza por cordones o nidos de epitelio escamoso multiestratificado dispuestos en empalizada. La presencia de calcificaciones, acumulaciones de queratina prominentes y contenido quístico oscuro, rico en colesterol, lípidos y metahemoglobina, es típica de esta variante.
El tipo papilar (15%) está relacionado con metaplasia de células adenohipofisarias primitivas11,12 y se presenta habitualmente con gran componente quístico. Histológicamente está compuesto por epitelio escamoso formador de papilas. En esta variante son muy infrecuentes las calcificaciones y la infiltración peritumoral13.
En más del 70% de las formas adamantinomatosas se detecta una mutación en el exón 3 del gen de la β-catenina que no está presente en el craneofaringioma de tipo papilar14.
DiagnósticoEl diagnóstico de sospecha del craneofaringioma se basa en los hallazgos clínicos y radiológicos, y se confirma con el estudio histológico.
Sospecha clínicaLa sintomatología más frecuente deriva de su naturaleza expansiva y/o del desarrollo de hipertensión intracraneal. Las alteraciones visuales se producen en el 50% de los casos, debido a la hipertensión intracraneal o al efecto de masa que inducen cambios en la vascularización de los nervios ópticos y pueden resultar en una pérdida de visión permanente. Las alteraciones de la función motora de pares craneales (II, IV y VI) se producen por la invasión de los senos cavernosos.
La afectación endocrina es mucho más evidente en la infancia, ya que la disfunción hipotalámico-hipofisaria afecta al desarrollo y al crecimiento normales3,15. Al ser tumores de crecimiento lento los síntomas son insidiosos e inespecíficos, lo que habitualmente retrasa el diagnóstico entre 1 y 2años.
En la infancia los síntomas/signos más frecuentes son la pérdida de visión, los síntomas derivados de la hipertensión intracraneal (irritabilidad, náuseas, vómitos, papiledema e incluso macrocefalia si las suturas craneales no están cerradas) y el fallo del crecimiento. En la preadolescencia destacan las alteraciones visuales (pérdida de agudeza visual y defectos campimétricos) y los trastornos del desarrollo puberal. En la edad adulta los síntomas más frecuentes son el hipogonadismo con disfunción eréctil y oligoamenorrea (45-65%) y las alteraciones visuales3,16-18. La hiperfagia y la obesidad, la poliuria y la polidipsia (diabetes insípida), asociadas a afectación hipotalámica importante, se presentan a cualquier edad (tabla 1).
Niveles de evidencia y grados de recomendación
| Niveles de evidencia |
| Ia. Evidencia obtenida de metaanálisis de ensayos clínicos de alta calidad (controlados y aleatorizados) |
| Ib. Evidencia obtenida de al menos un ensayo clínico de alta calidad |
| IIa. Evidencia obtenida de un estudio bien diseñado controlado sin aleatorizados |
| IIb. Evidencia obtenida de al menos un estudio bien diseñado casi experimental |
| III. Evidencia obtenida de estudios descriptivos bien diseñados y de estudios casos-controles |
| IV. Opinión de expertos |
| Grados de recomendación |
| A. Nivel de evidencia Ia o Ib |
| B. Nivel de evidencia IIa, IIb o III |
| C. Nivel de evidencia IV |
| Buena práctica clínica |
De Atkins et al.2.
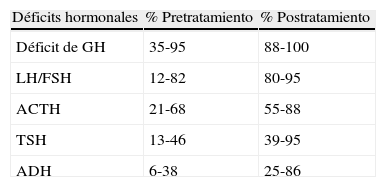
En las tablas 2 y 3 se describen las frecuencias de las distintas alteraciones hormonales3,5,19-21 y las manifestaciones no endocrinas3,5,22,23. Otras manifestaciones no endocrinas son los trastornos de la conducta y dificultades para la memoria y el aprendizaje24–27.
Manifestaciones no hormonales del craneofaringioma
| Derivadas de la hipertensión intracraneal (70-80%) |
| Cefalea |
| Náuseas, vómitos |
| Alteraciones visuales (50-70%) |
| Hemianopsia, escotomas y papiledema |
| Derivadas de la afectación hipotalámica (15-40%) |
| Hipotensión ortostática |
| Obesidad/hiperfagia |
| Hipo/hipertermia |
| SAOS |
| Otras (10-30%) |
| Epilepsia, meningitis química por rotura de quistes al espacio subaracnoideo |
| Alteraciones de la memoria a corto plazo y dificultades de concentración |
| Trastornos del sueño |
| Depresión, letargia |
Los craneofaringiomas miden generalmente más de 2cm (14-20% >4cm, 58-76% entre 2 y 4cm, y 4-28% <2cm)24,28 La imagen radiológica clásica es una masa sólida (18-39%) o quístico-sólida (46-64%) con diversos grados de calcificación (en palomitas de maíz o «en cáscara»). La hidrocefalia (20-38%) está presente sobre todo en niños (50% al diagnóstico). El diagnóstico más probable ante una lesión paraselar calcificada quística, incluso con una radiografía simple de cráneo, es el de craneofaringioma.
Mediante TAC se pueden demostrar claramente las calcificaciones características (90%) y evaluar el tamaño y las relaciones anatómicas del tumor, sobre todo cuando se emplean técnicas de alta resolución (TACAR).
La RM delimita, además del tamaño, la extensión tumoral, la infiltración de hipotálamo y tercer ventrículo y, a través de secuencias potenciadas en T1 y T2, el edema perineural, la gliosis reactiva y la invasión tumoral (línea de hiperintensidad parenquimatosa), siendo la prueba de elección para planificar el abordaje quirúrgico29-31.
Los tumores homogéneos, hipointensos en T1 (o hiperintensos si poseen metahemoglobina y material proteináceo) e hiperintensos en T2, que no se realzan tras contraste, indican una naturaleza fundamentalmente quística (colesterol, queratina). En T1 pueden mostrar una delgada línea periférica. En cambio, los tumores sólidos son heterogéneos iso o hipointensos en T1, y con realce tras gadolinio o en T2. Las calcificaciones pueden visualizarse como zonas de baja señal en T1 y T232.
En muchas ocasiones es conveniente completar el estudio con una angiorresonancia magnética (angioRM) y/o angioTC, que prácticamente han suplantado a la angiografía, para delinear el trayecto de los vasos y descartar aneurismas y malformaciones vasculares29. En algunos casos pueden ser de utilidad la ecografía transfontanela en el primer año de vida, y el PET/TC con metionina para el diagnóstico diferencial con otros tumores de la región y con la radionecrosis.
Evaluación endocrinaEs preciso realizar una evaluación hormonal completa hipotálamo-hipofisaria, normalmente mediante determinaciones basales (GH/IGF-1, LH, FSH/estradiol-testosterona, TSH/T4L, ACTH/cortisol). La determinación de la diuresis, iones y osmolaridad en plasma y orina permite la evaluación de la secreción de vasopresina. En la infancia se precisa una estimación de la edad ósea; en situación peripuberal con adelanto o retraso puberal puede además ser útil la ecografía ovárica.
Algunas alteraciones hormonales, como el hipocortisolismo, el hipotiroidismo y las alteraciones del equilibrio hidroelectrolítico, deben corregirse antes de la intervención quirúrgica33.
Evaluación oftalmológicaLa evaluación de la agudeza y el campo visual es necesaria previamente a la cirugía, ya que pueden condicionar la urgencia, el abordaje quirúrgico y la agresividad de la intervención. Debe ser completada con una adecuada visualización de los discos ópticos, para excluir papiledema, y la realización de unos potenciales visuales evocados o una tomografía de coherencia óptica (OCT) si está disponible para valorar la intensidad de la afectación de la vía óptica y su pronóstico de recuperación tras el tratamiento quirúrgico.
Recientemente se ha propuesto, en niños afectos de craneofaringioma, una escala de valoración preoperatoria de 5 áreas funcionales (cada una escalada en 4 grados): visual, neurológica, hipofisaria, hipotalámica y social, que permitiría una mejor predicción de los resultados postoperatorios y, por tanto, una mejor información y consejo médico para el paciente y su familia34.
TratamientoLa cirugía es el tratamiento de elección en la gran mayoría de los craneofaringiomas. El objetivo es establecer un diagnóstico histológico definitivo, reducir los síntomas compresivos y resecar la mayor parte de la masa tumoral con la menor morbilidad posible. Sin embargo, a pesar de los avances en las técnicas neuroquirúrgicas, los craneofaringiomas a menudo son difíciles de resecar completamente, ya que tienen márgenes irregulares, están adheridos a los tejidos adyacentes y con frecuencia son de gran tamaño en el momento del diagnóstico.
La elección de la vía de abordaje, así como la amplitud de la resección, continúan siendo temas de debate. Algunos grupos apoyan una cirugía agresiva con el objeto de conseguir la resección completa del tumor, mientras que otros optan por una cirugía más conservadora seguida de radioterapia para tratar la enfermedad residual. Las técnicas de radioterapia actuales limitan la exposición a la radiación ionizante de los tejidos adyacentes al tumor3,35, reduciendo los efectos secundarios. El desarrollo de las técnicas de cirugía y radioterapia, la adecuada sustitución hormonal, así como la mejora en el soporte médico perioperatorio, han alargado la supervivencia de estos pacientes. Por otra parte, el desarrollo frecuente de estos tumores en niños y jóvenes y su proximidad a la vía óptica, el polígono de Willis y el sistema hipotálamo-hipofisario nos obliga a tener en cuenta las posibles secuelas a largo plazo de las distintas opciones terapéuticas3,35.
CirugíaExtensión de la resecciónTradicionalmente, el objetivo de la cirugía ha sido la resección total o casi total (>95%) del tumor, principalmente por craneotomía. Sin embargo, los efectos secundarios derivados de esta estrategia, como la lesión hipotalámica, el deterioro cognitivo y el empeoramiento secundario de la calidad de vida, pueden apoyar el empleo de una resección más limitada seguida de radioterapia.
La posibilidad de resección completa se reduce en tumores mayores de 4cm, en los de localización retroquiasmática o con invasión del tercer ventrículo, con adherencia a las estructuras neurovasculares adyacentes o con calcificaciones en más del 10% del volumen tumoral, así como en los tumores asociados a hidrocefalia24.
En el 18-26% de los pacientes intervenidos con intención de realizar exéresis completa se comprueba radiológicamente la persistencia de restos tumorales36. Por otra parte, en las series con confirmación radiológica de resección completa las recidivas pueden producirse hasta en el 62% de los pacientes a los 10años de seguimiento, si bien en las series recientes el porcentaje de recidiva en estos casos es próximo al 25%25,37,38. Cuando la resección es parcial o subtotal (<95% del tumor) la frecuencia de recidiva es del 90-100% a los 10años; sin embargo, si se asocia radioterapia desciende hasta el 10-63% a los 10años, similar a la observada tras la resección total. En la serie de Rajan et al.39 la amplitud de la cirugía en pacientes posteriormente irradiados no fue un factor independiente de predicción de recurrencia.
La supervivencia libre de progresión tras cirugía radical es del 70% a los 10años, mientras que es solo del 9% tras cirugía subtotal sin radioterapia. La supervivencia global y la supervivencia libre de progresión son similares entre los pacientes intervenidos con intención radical y los tratados mediante cirugía subtotal seguida de radioterapia40,41. No obstante, hay que tener en cuenta que estos resultados pueden estar sesgados por la decisión de cirugía con intención radical en los tumores menos agresivos, con mayor probabilidad de curación solo con resección quirúrgica. Los peores resultados (menor supervivencia global y supervivencia libre de progresión) se obtienen con cirugía subtotal sin radioterapia adyuvante, con el 60-80% de recidiva durante el primer año tras la cirugía inicial21.
Vía de abordajeLa selección de la vía de abordaje depende de la localización, la consistencia, la morfología y el tamaño del tumor, de la presencia de calcificaciones y de la experiencia del cirujano.
En caso de hidrocefalia asociada, es necesario proceder a la descompresión de los ventrículos antes de la exéresis del tumor. De la misma forma, la evacuación del componente quístico cuando está presente, especialmente si es de gran tamaño, facilita la resección del componente sólido del tumor.
Es difícil comparar los resultados de una u otra técnica, dada la naturaleza retrospectiva de la mayoría de las series quirúrgicas y el número limitado de pacientes incluidos por cada centro. En un reciente metaanálisis que compara el abordaje transesfenoidal y transcraneal del craneofaringioma en niños, la vía transesfenoidal se asoció a mejor control tumoral y menor morbilidad posquirúrgica42. La incidencia de recidiva tras resección completa del tumor por vía transcraneal fue superior respecto a la vía transesfenoidal (17% frente al 8%). Sin embargo, los autores concluyen que estas diferencias pueden estar relacionadas con la elección de la vía transesfenoidal en tumores de menor tamaño y de predominio intraselar, mientras que los pacientes derivados a cirugía transcraneal tenían tumores de mayor tamaño, con predominio de crecimiento supraselar y en los que era más frecuente la hidrocefalia.
La elección de la vía de abordaje debe ser individualizada en función de las características del tumor, la edad del paciente, la intención radical o subtotal de la resección, así como la previsión de administrar o no radioterapia en caso de resección subtotal. La vía transesfenoidal, en los tumores accesibles mediante este abordaje, debería ser considerada en niños en que se planea resección subtotal seguida de radioterapia, ya que se asocia a menor morbilidad postoperatoria.
RadioterapiaLa radioterapia externa es el tratamiento estándar para el control de restos tumorales poscirugía o recidivas locales.
La dosis óptima de radiación no ha sido establecida, debido a la carencia de estudios aleatorizados controlados, si bien habitualmente se emplean 45-55Gy. En general se administra en fracciones diarias de 1,8-2Gy, 5días por semana durante 6semanas, para minimizar la toxicidad sobre la vía óptica. Algunos estudios relacionan la frecuencia de recidiva con la dosis de radioterapia poscirugía subtotal, mayor en los pacientes que reciben dosis inferiores a 5.000rads43. El control tumoral a 10años tras el tratamiento con cirugía y radioterapia varía entre el 57 y el 89%, mientras que es del 31-42% tras solo cirugía.
Las técnicas modernas de radioterapia, como la radioterapia estereotáxica fraccionada o la radiocirugía, permiten la aplicación de altas dosis de radioterapia a una diana definida, reduciendo el daño de los tejidos adyacentes.
Radiocirugía estereotáxicaSu utilidad en el tratamiento del craneofaringioma ha sido evaluada en algunas series con un tiempo de seguimiento limitado44,45. En la mayoría de casos se utilizó como tratamiento adyuvante ante la persistencia o la recidiva tumoral tras cirugía con o sin radioterapia convencional previa, mientras que se empleó como tratamiento primario en un menor número de casos. Globalmente se consiguió control tumoral en el 90% de los tumores sólidos y en el 60% de los tumores mixtos44,46. En la serie de Kobayashi47, que reúne 98 pacientes, se consiguió el control tumoral en un 80% de los casos, con respuesta completa en el 20%. En las series que incluyeron la estabilización del tumor como criterio de respuesta, esta ascendió al 85%. La supervivencia libre de progresión a los 5 y 10años fue del 60,8 y del 53,8%, respectivamente. La respuesta fue mejor en los tumores <2cm de diámetro y peor en los tumores quísticos, que a menudo requirieron tratamiento específico del quiste, como aspiración, irradiación o quimioterapia intracavitaria. La frecuencia de neuropatía óptica fue del 1,8% en los pacientes que recibieron <10Gy y 6,9% en los que recibieron dosis superiores.
La principal indicación de la radiocirugía estereotáxica es el tratamiento de lesiones sólidas de pequeño tamaño (<3cm) y bien definidas, que distan al menos 2mm de la vía óptica, residuales o recurrentes tras cirugía y/o radioterapia convencional.
Radioterapia estereotáxica fraccionadaLa radioterapia estereotáxica fraccionada combina la precisión en la selección de la zona a tratar con el fraccionamiento de la dosis total de radiación, reduciendo la irradiación del tejido normal y, por tanto, los efectos a largo plazo de la radioterapia. Ha sido evaluada en una serie limitada de casos de craniofaringioma con buenos resultados. En la mayoría de ellos se ha utilizado como tratamiento adyuvante tras cirugía en pacientes con restos tumorales residuales o recurrencias. En las series publicadas el control tumoral es excelente, con una supervivencia libre de progresión de 92-100% a los 5años48-50.
En un reciente metaanálisis de los estudios publicados sobre el tratamiento del craneofaringioma con radiocirugía o radioterapia estereotáxica fraccionada en un total de 252 pacientes, el control tumoral se alcanza en el 69% de los casos. Tras un seguimiento medio de 57 meses, describen efectos secundarios endocrinológicos o neurológicos en el 0-34%48. En un estudio de 50 pacientes tratados con radiocirugía o radioterapia estereotáxica fraccionada el control tumoral fue similar entre las 2 técnicas. La radiocirugía no se asoció a menos efectos secundarios26.
Controversias respecto al momento de la irradiaciónExiste controversia respecto al momento de administrar radioterapia. En algunas series que comparan la eficacia de la radioterapia (radiocirugía o radioterapia estereotáxica fraccionada) si se administra inmediatamente tras la cirugía al comprobarse persistencia de restos tumorales (radioterapia adyuvante), respecto a su administración cuando se comprueba crecimiento de los restos o recurrencia del tumor (radioterapia de rescate), no se observan diferencias en la supervivencia libre de progresión26,50. Por otra parte, dado que la irradiación puede asociarse a disfunción hipotálamo-hipofisaria y a alteración visual, y que dificulta el tratamiento quirúrgico de las recidivas, los autores defienden su empleo cuando se demuestre radiológicamente recidiva o crecimiento de restos tumorales, con el objeto de reducir la morbilidad asociada a radioterapia. Sin embargo, Lin et al.27 recomiendan remitir a radioterapia tras cirugía a los pacientes con elevada probabilidad de recurrencia, ya que obtienen mejor control local sin aumento en la incidencia de complicaciones. Los efectos secundarios de la radioterapia han de valorarse con especial cuidado en niños menores de 5años, y debe valorarse el riesgo de futuras intervenciones terapéuticas en los casos de alto riesgo de recidiva.
Quimioterapia sistémicaLa eficacia de la quimioterapia en el craneofaringioma ha sido evaluada en un número pequeño de casos, principalmente niños y adultos jóvenes con tumores progresivos o recurrentes. Se ha utilizado doxorubicina, con control de la enfermedad local en 3 de 4 niños tratados51. En una serie de 12 jóvenes, el interferón alfa solo redujo el volumen tumoral en 3 de ellos, si bien al suspender el tratamiento se observó crecimiento tumoral en el 67% de los casos que habían completado un año de terapia sin progresión52.
Tratamiento de las lesiones quísticasEl 90% de los craneofaringiomas tienen un componente quístico, que en ocasiones constituye el principal componente del tumor y requiere un tratamiento específico.
Vaciamiento del quisteLas lesiones recurrentes con importante componente quístico que no son susceptibles de exéresis completa pueden ser tratadas mediante aspiración intermitente por punción estereotáxica o a través de un reservorio tipo Ommaya53 o bien mediante cistostomía ventrículo-cisternal, con evacuación del contenido del quiste al líquido cefalorraquídeo.
Irradiación intracavitariaConsiste en la instilación estereotáxica de isótopos que emiten radiaciones β. La dosis de radiación sobre las células del epitelio secretor que recubre las paredes del quiste es superior a la obtenida con radioterapia externa, reduciendo la producción de fluido y el volumen del quiste. Los isótopos más utilizados son el 90itrio, el 32fósforo y el 196renio. La radiación administrada a la pared del quiste varía entre 200 y 267Gy54. La respuesta inicial es buena, con reducción en el tamaño del quiste en más del 70% de los casos, principalmente durante los 2 primeros años. Sin embargo, el 33% recidivan por incremento del tamaño del quiste tratado, por la aparición de nuevos quistes o por aumento de la porción sólida del tumor, y el 54% requieren tratamientos adicionales55,56. En el 5% de los casos puede producirse pérdida visual y radionecrosis de las zonas próximas.
Quimioterapia intracavitariaConsiste en la instilación de un agente antineoplásico (principalmente bleomicina, y más recientemente interferón) dentro de la cavidad del quiste a través de un reservorio tipo Ommaya conectado a un catéter. El tratamiento endocavitario con bleomicina estaría indicado en el manejo de craneofaringiomas predominantemente quísticos en los que no es posible la resección completa y se prefiere retrasar la radioterapia. La reducción en el volumen del quiste puede permitir la resección del tumor. La experiencia con IFNα endocavitario es menor que con la bleomicina. Al igual que esta, puede permitir el control a corto plazo de un craneofaringioma principalmente quístico y retrasar el tratamiento definitivo asociado a mayor morbilidad, lo cual puede ser importante en los niños de menor edad. Por otra parte, el IFNα es menos tóxico que la bleomicina si se extravasa al espacio subaracnoideo57.
RecidivasLa mayoría de las recidivas se diagnostican durante los primeros 3 o 4años tras el tratamiento inicial. El principal factor de riesgo de recurrencia es la presencia de restos tumorales tras la primera intervención. La radioterapia, precedida o no de una segunda cirugía, es muy útil en el control de la masa tumoral.
El tratamiento quirúrgico de la recidiva consigue la exéresis completa en menos del 25% de los casos en la mayoría de las series, debido a la presencia de adherencias y fibrosis secundarias a los tratamientos previos, que dificultan la resección. Sin embargo, en algunas series el porcentaje de pacientes en que se consigue la resección total es elevado (61-79%), con aceptable morbilidad posquirúrgica34,58. Los autores atribuyen parte de sus buenos resultados a un seguimiento radiológico estrecho de los pacientes, que les permite detectar recidivas en una fase temprana. En general, la mortalidad y la morbilidad (10-24%) son mayores respecto a la cirugía del tumor primario, por lo que con frecuencia la resección del tumor será paliativa.
Respecto a la radioterapia, la supervivencia libre de progresión a los 10años desde la recurrencia es del 72%. Estos resultados son similares en los pacientes que previamente a la radioterapia han sido sometidos a una segunda intervención y en los que solo recibieron radioterapia al demostrarse recidiva posterior a la primera cirugía59.
Morbilidad asociada al tumor y su tratamientoLas secuelas secundarias al daño tisular producido por el tumor primario o recurrente, así como por su tratamiento, incluyen alteraciones en la función hipotalámica, secreción hormonal, alteraciones visuales, así como trastornos del comportamiento y de la función cognitiva, que tienen un gran impacto en la calidad de vida del paciente y su integración psicosocial. Los efectos a largo plazo de la radioterapia se asocian a la dosis total y la administrada por fracción, el volumen de tejido normal expuesto a la radiación y la edad del paciente, siendo mayores las secuelas en niños. Posiblemente, el empleo de nuevas técnicas de radioterapia y la administración de dosis inferiores a 55Gy o 1,8Gy por fracción producirá menor toxicidad a largo plazo.
Disfunción endocrinológicaTras la cirugía, el déficit hormonal asciende al 73-100% de los pacientes, y entre el 54-100% de los pacientes tienen al menos déficit de 3 hormonas (tabla 2).
La incidencia de hipopituitarismo tras la radioterapia es difícil de valorar, ya que la mayoría de las series incluyen los pacientes tratados previamente con cirugía y el número de casos tratados solo con radioterapia es pequeño. En estas series la incidencia de panhipopituitarismo tras radioterapia oscila entre el 80 y el 100%60.
En los pacientes intervenidos se debe reevaluar la función hipotálamo-hipofisaria, mediante la determinación de GH/IGF-1, LH, FSH, estradiol-testosterona, TSH/T4L, ACTH/cortisol, diuresis, osmolaridad en plasma y orina, así como tests dinámicos en los casos en que se considere necesario. En pacientes sometidos a radioterapia deberá realizarse evaluación hormonal con periodicidad semestral para detectar nuevos déficits hormonales.
Disfunción hipotalámicaLa afectación hipotalámica secundaria al tumor o a su tratamiento puede producir hiperfagia y obesidad, trastornos en la regulación de la sed y el balance hidroelectrolítico, trastornos del comportamiento y la función cognitiva, alteraciones en el control de la temperatura, así como excesiva somnolencia diurna. Los factores que se asocian a mayor riesgo de disfunción hipotalámica son la presentación en niños de menor edad, presencia de disfunción hipotalámica desde el diagnóstico, invasión hipotalámica, tumor mayor de 3,5cm de diámetro, cirugía agresiva para resecar la totalidad del tumor o cirugía repetida por recurrencias, así como dosis de radiación superior a 51Gy60.
La obesidad está presente en el 26-61% de los pacientes intervenidos con o sin radioterapia adyuvante y es de origen multifactorial. El tumor y su tratamiento dañan los centros que controlan la saciedad y el balance energético en el suelo del tercer ventrículo. La lesión del núcleo ventromedial conduce a hiperfagia y obesidad, y la menor sensibilidad de las estructuras hipotalámicas a la leptina endógena estimula el apetito. La hiperinsulinemia favorecida por el desequilibrio autonómico con aumento en la actividad vagal favorece la adipogénesis. Por otra parte, en algunos estudios se ha demostrado que la ingesta calórica es similar a la de niños controles, por lo que los autores atribuyen la obesidad a la reducción de la actividad física, mediada en gran medida por las secuelas neurológicas, el déficit visual y la somnolencia diurna61,62. Tras la cirugía inicial todos los pacientes, incluidos aquellos con peso normal pero con daño hipotalámico debido al tumor o a su tratamiento, deberían ser incluidos en un programa de dieta y actividad física63.
El trastorno en la percepción de la sed está presente en el 14-19% de los pacientes intervenidos con o sin radioterapia adyuvante. Cuando se asocia a diabetes insípida aumenta notablemente el riesgo de deshidratación hipertónica y dificulta su manejo, requiriendo, además de la administración de desmopresina, un estricto control del balance hidroelectrolítico61.
Disfunción visualLa afectación visual está presente al diagnóstico en el 42% de los casos. Tras la cirugía mejora en el 46-66% de los casos en función de la extensión de la resección. Su frecuencia aumenta tras el tratamiento de las recidivas62. Se ha asociado a la radioterapia cuando la dosis diaria en el fraccionamiento excede 2Gy.
Trastorno neurocognitivoEl deterioro en la función cognitiva y sus repercusiones psicosociales se han puesto de manifiesto en diversas series que evalúan los déficits motor y visual, el grado de dependencia para las actividades diarias, la capacidad para hacer el trabajo que realizaban previamente, el rendimiento escolar o la capacidad para realizar su actividad cotidiana. En este sentido, los pacientes tratados con cirugía con o sin radioterapia tienen una deterioro psicosocial significativo hasta en el 47% de los casos durante el seguimiento. Además, el 50% tienen problemas de concentración, cambios de comportamiento, pérdida de memoria a corto plazo, anosmia o epilepsia63.
La evolución es peor en pacientes con tumores grandes o asociados a hidrocefalia, o en los sometidos a una segunda cirugía por recidiva o progresión del crecimiento del tumor. En algunas series los pacientes sometidos a cirugía conservadora con radioterapia adyuvante tenían mejor rendimiento escolar y recuperaban con mayor frecuencia el empleo que los sometidos solo a cirugía. No obstante, hay que tener en consideración los posibles efectos secundarios a largo plazo de la radioterapia, si bien las nuevas técnicas de irradiación pueden reducir el riesgo de efectos adversos64.
La influencia de la opción terapéutica sobre la función cognitiva es difícil de evaluar ante la ausencia de estudios prospectivos que incluyan una valoración neuropsicológica antes y después del tratamiento. La inclusión de estos tests en la evaluación de los pacientes puede ayudar evaluar el impacto del tratamiento en la calidad de vida.
MortalidadLa mortalidad en el craneofaringioma es 3-6 veces superior a la de la población general, y también superior a la referida en otras causas de hipopituitarismo65. Los avances en las técnicas de neurocirugía, radioterapia y de soporte perioperatorio, incluido el tratamiento sustitutivo del déficit suprarrenal y la diabetes insípida, han mejorado la supervivencia respecto a décadas anteriores. Estudios recientes indican una supervivencia a los 10años de seguimiento de entre el 83 y el 93%5.
Respecto a la modalidad de tratamiento, algunas series no encuentran diferencias significativas en la supervivencia a los 10años entre los pacientes sometidos a cirugía con resección completa (62-100%), parcial (86%) o con radioterapia adyuvante (74-100%). Sin embargo, la resección parcial implica mayor frecuencia de recidiva y necesidad de reintervención y/o radioterapia, con el consiguiente incremento en la morbilidad asociada64. La mortalidad perioperatoria actual en la cirugía inicial oscila entre el 1,7 y el 5,4%. Sin embargo en las reintervenciones por recidiva la mortalidad asciende al 2,2-24%34. Los factores de riesgo de reducción en la supervivencia son la resección incompleta, el tamaño del tumor superior a 5cm y la presencia de hidrocefalia o derivación ventrículo-peritoneal.
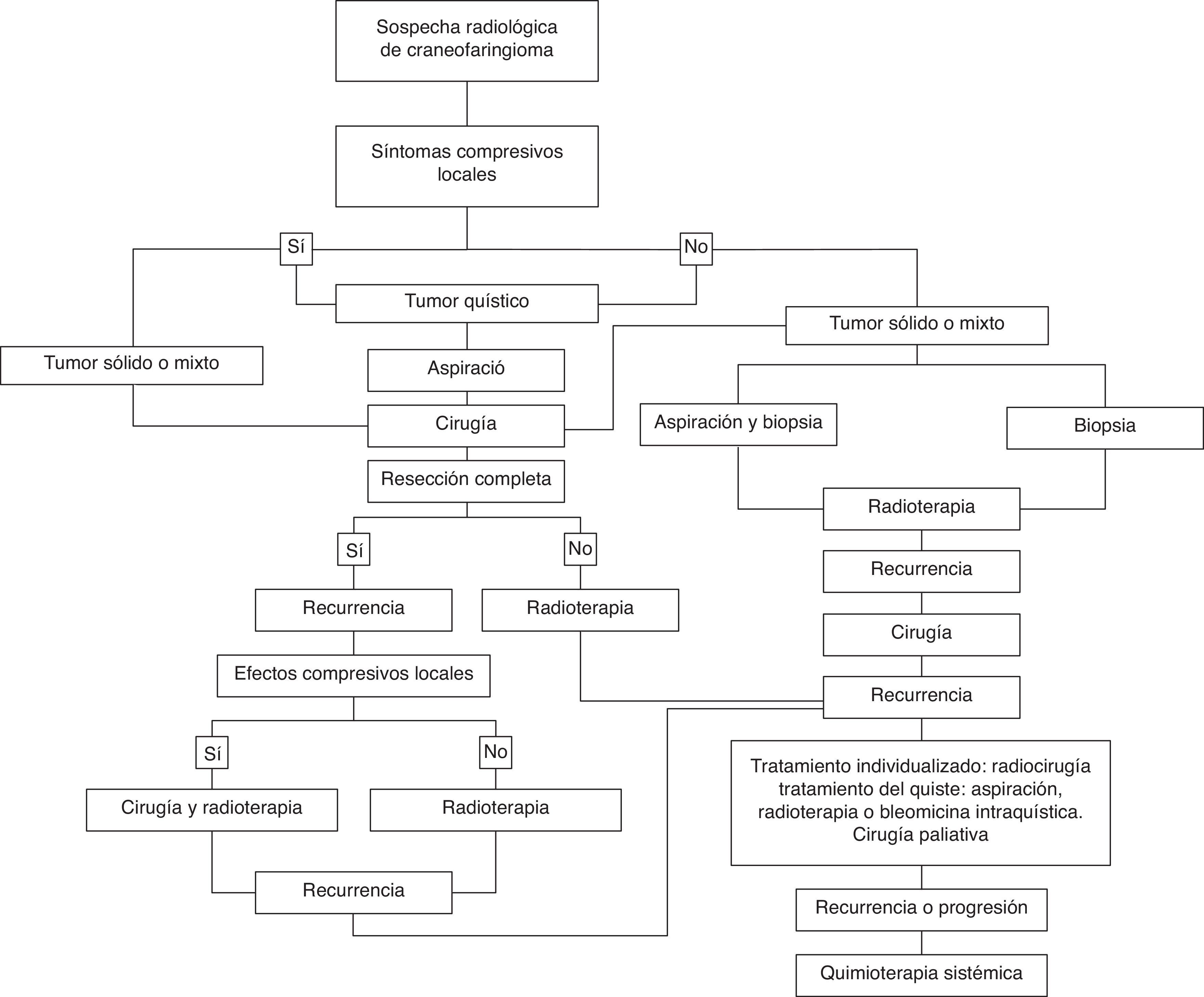
Recomendaciones de tratamiento basadas en la evidenciaAnte una lesión radiológicamente sugestiva de craneofaringioma está indicada la intervención terapéutica. En la figura 1 se propone un algoritmo de tratamiento.

La cirugía es el tratamiento primario de elección en la mayoría de los casos, y muy especialmente en aquellos en que se asocia sintomatología compresiva local, permitiendo además la confirmación histológica del tumor (grado de recomendación B, nivel de evidencia IIa).
Si es posible, se intentará la resección completa de la lesión (grado de recomendación B, nivel de evidencia IIa). No obstante, en aquellos casos en que por las características del tumor sea previsible una morbilidad importante, principalmente hipotalámica y/o la persistencia de restos, es aceptable una resección parcial o subtotal seguida de radioterapia (grado de recomendación B, nivel de evidencia IIa). La vía de abordaje, transcraneal a transesfenoidal, se elegirá de forma individualizada en función de la localización, el tamaño y la consistencia del tumor.
Si tras la cirugía se comprueba la persistencia de restos tumorales se recomienda completar el tratamiento con radioterapia, dada la alta frecuencia de recrecimiento del tumor y la mayor morbilidad asociada a la reintervención (grado de recomendación B, nivel de evidencia III). No obstante, el momento de la irradiación, inmediatamente tras la resección subtotal o al detectarse crecimiento de los restos tumorales, es controvertido, especialmente en niños. Las nuevas técnicas de radioterapia pueden minimizar la toxicidad del tratamiento y propiciar su empleo más precozmente.
Por otra parte, los tumores pequeños no asociados a síntomas compresivos locales, pueden ser tratados solo con radioterapia, previa confirmación histológica del tumor mediante biopsia. En general esta opción se reserva para los pacientes que son malos candidatos quirúrgicos.
Los craneofaringiomas con gran componente quístico deberían ser evacuados previamente a la cirugía para facilitar su resección. También deben ser evacuados antes de la radioterapia, ya que esta puede producir un crecimiento transitorio del quiste y síntomas compresivos locales.
Las lesiones recurrentes previamente intervenidas deberían ser evaluadas de forma individual. Si es posible la resección total o parcial de la recidiva con una morbilidad aceptable, estaría indicada la cirugía, especialmente en pacientes con síntomas compresivos locales (grado de recomendación B, nivel de evidencia IIa). En el resto de los casos estaría indicada la radioterapia, utilizando preferiblemente radioterapia estereotáxica fraccionada para minimizar los efectos secundarios de la radiación (grado de recomendación B, nivel de evidencia III). La radiocirugía debería considerarse en función de las características del tumor, especialmente en el caso de recidiva en pacientes que han sido sometidos a radioterapia convencional previa.
Si la lesión recurrente tiene un componente quístico significativo, la aspiración de su contenido y la administración de radioterapia o quimioterapia intracavitaria puede ser de utilidad.
Lesiones paraselaresSon lesiones de prevalencia muy baja. Son escasos los estudios sistemáticos de cada una de ellas, aunque dada su rareza no es difícil encontrar publicaciones de casos aislados. La región paraselar es un área anatómica compleja en la que se pueden desarrollar una serie de enfermedades neoplásicas, inflamatorias, infecciosas, embrionarias y vasculares (tabla 4). El diagnóstico de estas lesiones supone un enfoque multidisciplinario y es esencial, ya que el tratamiento de elección será diferente para cada trastorno66. En la mayoría de los casos el clínico las diagnostica basándose en el informe anatomopatológico de un caso quirúrgico. Las pruebas funcionales hipofisarias no ayudan al diagnóstico diferencial. Este puede alcanzarse, en ocasiones, por el contexto clínico o por TC, RM y otras técnicas de imagen67. La gammagrafía con el isótopo 99mTc(V)-DMSA puede llegar a ser útil para diferenciar lesiones paraselares de los adenomas hipofisarios68 La TC es particularmente útil para identificar cambios óseos y calcificaciones intra y perilesionales. La RM es la técnica de elección por su gran sensibilidad y alta resolución espacial que permite separar el tejido normal del patológico, definiendo sus componentes sólidos, quísticos, hemorrágicos o lipídicos67. La arteriografía por sustracción digital ayuda en lesiones vasculares y tumores muy vascularizados. En pacientes seleccionados se puede plantear biopsia endoscópica transnasal; sin embargo, el riesgo/beneficio es en la mayoría de los casos inasumible66. El comportamiento morfológico de las diferentes lesiones paraselares se detalla en la tabla 5.
Clasificación de las lesiones paraselares (no adenoma hipofisario) según su topografía y frecuencia
| Supraselares |
| Más frecuentes |
| Craneofaringioma |
| Glioma óptico |
| Glioma hipotalámico |
| Germinoma |
| Astrocitoma |
| Metástasis |
| Aneurismas/fístulas arteriovenosas |
| Menos frecuentes |
| Quiste de Ratkhe |
| Quiste aracnoideo |
| Coristoma |
| Granuloma idiopático |
| Tuberculosis |
| Sarcoidosis |
| Histiocitosis |
| Dermoide |
| Hamartoma del tuber cinereum |
| Lateroselares |
| Más frecuentes |
| Meningioma |
| Schwannoma |
| Traumatismo |
| Menos frecuentes |
| Síndrome de Tolosa-Hunt |
| Aneurisma intracavernoso |
| Cordoma |
| Carcinoma nasofaríngeo |
| Metástasis de la base del cráneo |
| Linfoma |
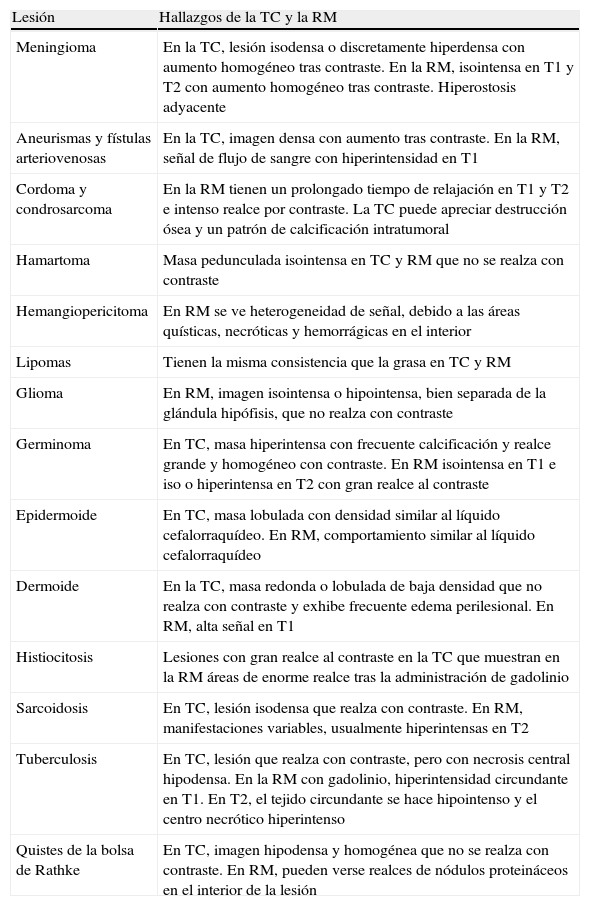
Comportamiento morfológico de las lesiones paraselares
| Lesión | Hallazgos de la TC y la RM |
| Meningioma | En la TC, lesión isodensa o discretamente hiperdensa con aumento homogéneo tras contraste. En la RM, isointensa en T1 y T2 con aumento homogéneo tras contraste. Hiperostosis adyacente |
| Aneurismas y fístulas arteriovenosas | En la TC, imagen densa con aumento tras contraste. En la RM, señal de flujo de sangre con hiperintensidad en T1 |
| Cordoma y condrosarcoma | En la RM tienen un prolongado tiempo de relajación en T1 y T2 e intenso realce por contraste. La TC puede apreciar destrucción ósea y un patrón de calcificación intratumoral |
| Hamartoma | Masa pedunculada isointensa en TC y RM que no se realza con contraste |
| Hemangiopericitoma | En RM se ve heterogeneidad de señal, debido a las áreas quísticas, necróticas y hemorrágicas en el interior |
| Lipomas | Tienen la misma consistencia que la grasa en TC y RM |
| Glioma | En RM, imagen isointensa o hipointensa, bien separada de la glándula hipófisis, que no realza con contraste |
| Germinoma | En TC, masa hiperintensa con frecuente calcificación y realce grande y homogéneo con contraste. En RM isointensa en T1 e iso o hiperintensa en T2 con gran realce al contraste |
| Epidermoide | En TC, masa lobulada con densidad similar al líquido cefalorraquídeo. En RM, comportamiento similar al líquido cefalorraquídeo |
| Dermoide | En la TC, masa redonda o lobulada de baja densidad que no realza con contraste y exhibe frecuente edema perilesional. En RM, alta señal en T1 |
| Histiocitosis | Lesiones con gran realce al contraste en la TC que muestran en la RM áreas de enorme realce tras la administración de gadolinio |
| Sarcoidosis | En TC, lesión isodensa que realza con contraste. En RM, manifestaciones variables, usualmente hiperintensas en T2 |
| Tuberculosis | En TC, lesión que realza con contraste, pero con necrosis central hipodensa. En la RM con gadolinio, hiperintensidad circundante en T1. En T2, el tejido circundante se hace hipointenso y el centro necrótico hiperintenso |
| Quistes de la bolsa de Rathke | En TC, imagen hipodensa y homogénea que no se realza con contraste. En RM, pueden verse realces de nódulos proteináceos en el interior de la lesión |
La región paraselar no está claramente definida e incluye todas las estructuras que rodean la silla turca. Pueden resultar involucradas estructuras vitales como el parénquima cerebral, las meninges, los nervios ópticos y otros nervios craneales, los vasos sanguíneos y compartimentos óseos. Las lesiones paraselares se presentan habitualmente con hipopituitarismo o síntomas de efecto de masa debido a la compresión de las estructuras cercanas. Su gravedad depende de la localización, el tamaño y el potencial de crecimiento de los tumores. La pérdida de visión es un motivo de consulta frecuente, y el 25% de los casos desarrollan alteraciones de los pares craneales. La diabetes insípida es un hallazgo común en los tumores paraselares66. La clasificación de la lesiones paraselares basada en su topografía (supraselar o lateroselar) es útil, pues puede tener repercusión sintomática. En las lesiones supraselares son más frecuentes la clínica hipotalámica y la disfunción hormonal por interferencia con el tallo hipofisario. Las lateroselares pueden expresarse por síntomas neurológicos relativos a la afectación de la vía óptica, el seno cavernoso o el polígono de Willis. Por eso se propone la clasificación modificada de Ruscalleda67 que se muestra en la tabla 4.
El comportamiento morfológico de las diferentes lesiones paraselares3 se detalla en la tabla 5.
La cirugía es el tratamiento de elección en la mayoría de los casos. Las posibles vías de abordaje son la transesfenoidal, la transcraneal y recientemente la cirugía endonasal endoscópica extendida. La radioterapia y los tratamientos médicos pueden ser usados en función del tipo de lesión que se diagnostique. En cuanto al pronóstico, depende del estado del paciente, de las enfermedades concomitantes, del tamaño del tumor y de la extensión, y de la histopatología del mismo. El seguimiento debe incluir evaluación de la función hormonal hipofisaria y técnicas de imagen.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
La finalidad de esta guía es la de ofrecerle un mejor conocimiento sobre algunos aspectos del craneofaringioma. La información contenida está escrita de manera genérica. Debido a esto, no todo lo detallado le será de utilidad para su caso particular.
Tenemos la esperanza que esta guía le ayude a entender esta enfermedad y le brinde una base para el diálogo con su médico o equipo de especialistas.
El cuerpo está formado por millones de células. Estas células se unen y crecen para crear los órganos y músculos del cuerpo. Sin embargo, algunas veces y por razones desconocidas, estas células se unen para crear un bulto o protuberancia sin función alguna en el cuerpo. Esta protuberancia es un tumor.
El craneofaringioma es un tumor benigno que se desarrolla cerca de la glándula pituitaria (una pequeña glándula endocrina ubicada en la base del cerebro). A diferencia del cáncer, el craneofaringioma no se extiende a otras partes del cuerpo.
Se sabe que esta afección no es hereditaria. Tampoco es el efecto de fármacos o de alguna enfermedad durante el embarazo.
El craneofaringioma produce síntomas de 3 formas diferentes:
- •
Aumento de la presión dentro del cerebro.
- •
Alteración de la función de la glándula pituitaria.
- •
Lesión del nervio óptico.
El incremento de la presión en el cerebro produce dolor de cabeza, náuseas, vómitos (especialmente en la mañana) y dificultades con el equilibrio.
La lesión de la glándula pituitaria produce desequilibrios hormonales que provocan sed y necesidad de orinar frecuentemente, retraso o adelanto en el desarrollo de la pubertad y trastornos del crecimiento en niños. En adultos puede producir deficiencias hormonales que pueden manifestarse como trastornos menstruales, disfunción sexual, cansancio, mareos, falta de apetito e hipotensión, entre otros.
Cuando el tumor daña el nervio óptico, se desarrollan problemas de visión.
La cirugía es el tratamiento principal para el craneofaringioma. El objetivo es quitar todo o la mayor parte del tumor. El tipo de cirugía puede variar en función del tamaño y de la localización del tumor:
Cirugía transesfenoidal. En caso de que el tumor sea pequeño y localizado dentro de la silla turca, este podrá ser extirpado a través de los orificios de la nariz o del labio superior.
Craneotomía. En caso de que el tumor sea de mayor tamaño o esté localizado por encima de la silla turca, se accederá a través del cráneo.
En tumores que no se pueden extirpar totalmente solo con cirugía, por lo general, es necesaria la radioterapia para prevenir que se reproduzca el tumor.
Si el tumor tiene un importante componente quístico, puede ser necesario aspirar su contenido a través de un orificio en el cráneo para drenar la sustancia del tumor.
El cerebro tiene muchas funciones importantes, como la regulación del apetito, del sueño y de la sed. Habitualmente estas funciones están a cargo de ciertas partes del cerebro que son muy delicadas. El craneofaringioma se desarrolla en zonas próximas a estas partes sensibles del cerebro.
Este tumor también es «pegajoso» y se adhiere fácilmente a los tejidos que lo rodean. Por este motivo, el tratamiento quirúrgico para extirpar el craneofaringioma sin dañar partes del cerebro es una labor delicada.
El pronóstico depende de si el tumor se puede extraer totalmente con cirugía o requiere radioterapia. El pronóstico para un paciente individual también depende de los problemas neurológicos y desequilibrios hormonales causados por el tumor y por el tratamiento. La mayoría de los problemas hormonales se pueden tratar satisfactoriamente con la medicación; sin embargo, los problemas visuales a veces no mejoran suficientemente con el tratamiento.
El craneofaringioma, así como el procedimiento quirúrgico necesario para su extracción o la radioterapia, pueden dañar el hipotálamo y la glándula hipofisaria.
En el hipotálamo se produce una hormona, llamada vasopresina, que actúa en el riñón regulando la retención o la eliminación del agua. Sin esta hormona, el cuerpo no retendrá fluidos y esto provocará la eliminación de gran cantidad de orina y sed.
La incapacidad del cerebro para producir vasopresina resulta en una afección llamada diabetes insípida. Sin tratamiento, esta afección puede provocar una severa deshidratación.
El tratamiento de la diabetes insípida consiste en suministrar al cuerpo una forma sintética de vasopresina llamada DDAVP (desmopresina). Esta se puede dar en comprimidos, en gotas intranasales o en espray.
Es importante no exceder la dosis recomendada. La sobredosis puede provocar una acumulación excesiva de líquido en el cuerpo y convulsiones. Por otro lado, si las dosis son bajas no se podrá controlar la diabetes insípida y aparecerá sed excesiva y necesidad de orinar frecuentemente.
Otras hormonas que pueden estar alteradas antes o después del tratamiento:
- •
Hormonas tiroideas (tiroxina). Estas hormonas son necesarias para el crecimiento y el metabolismo.
- •
Hormona del crecimiento. Usualmente esta hormona se administra después del tratamiento para normalizar el crecimiento en niños si es necesario.
- •
Hormonas sexuales. Estas hormonas pueden ser necesarias cuando la pubertad no empieza o si esta se desarrolla lentamente como consecuencia del tumor o su tratamiento. En los pacientes adultos son necesarias para mantener adecuadamente el aspecto masculino/femenino y la función sexual.
Los siguientes tratamientos pueden ser necesarios:
- •
Anticonvulsionantes. Se utilizan para prevenir las convulsiones.
- •
Corticosteroides. Se suministran altas dosis de dexametasona antes y después de la cirugía para prevenir o reducir la inflamación. Posteriormente los esteroides se emplean si faltan los que produce el propio organismo.
Los problemas de visión pueden mejorar después de la cirugía. Sin embargo, estos problemas también pueden ser permanentes. Por este motivo, será necesario evaluar la visión periódicamente.
También es importante llevar un registro de la estatura y del peso del individuo. Adicionalmente, se vigilará el desarrollo de la pubertad en los niños y adolescentes.
Finalmente, será necesario realizar analítica hormonal y un escáner o resonancia del cerebro periódicamente.
Estos efectos son el producto del daño causado al cerebro. Algunos de estos efectos pueden persistir a pesar de que el tumor ya no exista.
- •
Mayor ingesta de alimentos y obesidad.
- •
Dificultades para conciliar el sueño o somnolencia diurna.
- •
Deterioro en la capacidad de notar la sed.
- •
Problemas de memoria.
- •
Incapacidad para controlar la temperatura corporal.
La intención de esta guía es la de ofrecer una visión básica sobre el craneofaringioma. Puede consultar con su médico o equipo de especialistas en su localidad para mayor información.
