


INTRODUCCIÓN
El hipoparatiroidismo primario familiar es una entidad poco frecuente, que puede presentarse con un patrón de herencia autosómico dominante, autosómico recesivo o ligado al cromosoma X. Aunque la tetania y las convulsiones son las manifestaciones neurológicas más comúnmente asociadas, se han descrito casos de extrapiramidalismo relacionados con calcificaciones de los ganglios basales secundarias a alteraciones del metabolismo del calcio y fósforo en pacientes con hipoparatiroidismo.
La tríada de hipoparatiroidismo primario familiar, sordera neurosensorial y displasia renal con un patrón de herencia autosómico dominante, constituye una entidad clínica propia poco frecuente, que se asocia a anomalías cromosómicas concretas en algunos casos.
CASO CLÍNICO
Varón de 47 años de edad con rigidez, alteración de funciones superiores e incapacidad para caminar e interactuar con el medio. Tras un embarazo y un parto normales, el paciente debutó a partir de su primer año de vida con crisis generalizadas tonicoclónicas y retraso psicomotor leve. Fue ingresado en un colegio de educación especial, persistiendo únicamente cierta dificultad para la expresión oral. A los 18 años presentó deterioro de la marcha, debilidad, alopecia, disminución del nivel de conciencia en relación con procesos febriles y comportamiento incoherente, detectándose cifras de calcio sérico corregido de 5,9 mg/dl (valores normales: 8,5-10,5 mg/dl) y fósforo de 6,2 mg/dl (cifras normales: 2,5-4,5 mg/dl) con valores de paratohormona (PTH) menores de 3 pg/ml (10-65 pg/ml). Se le diagnosticó hipoparatiroidismo primario. En ese momento, el resto del estudio hormonal fue normal.
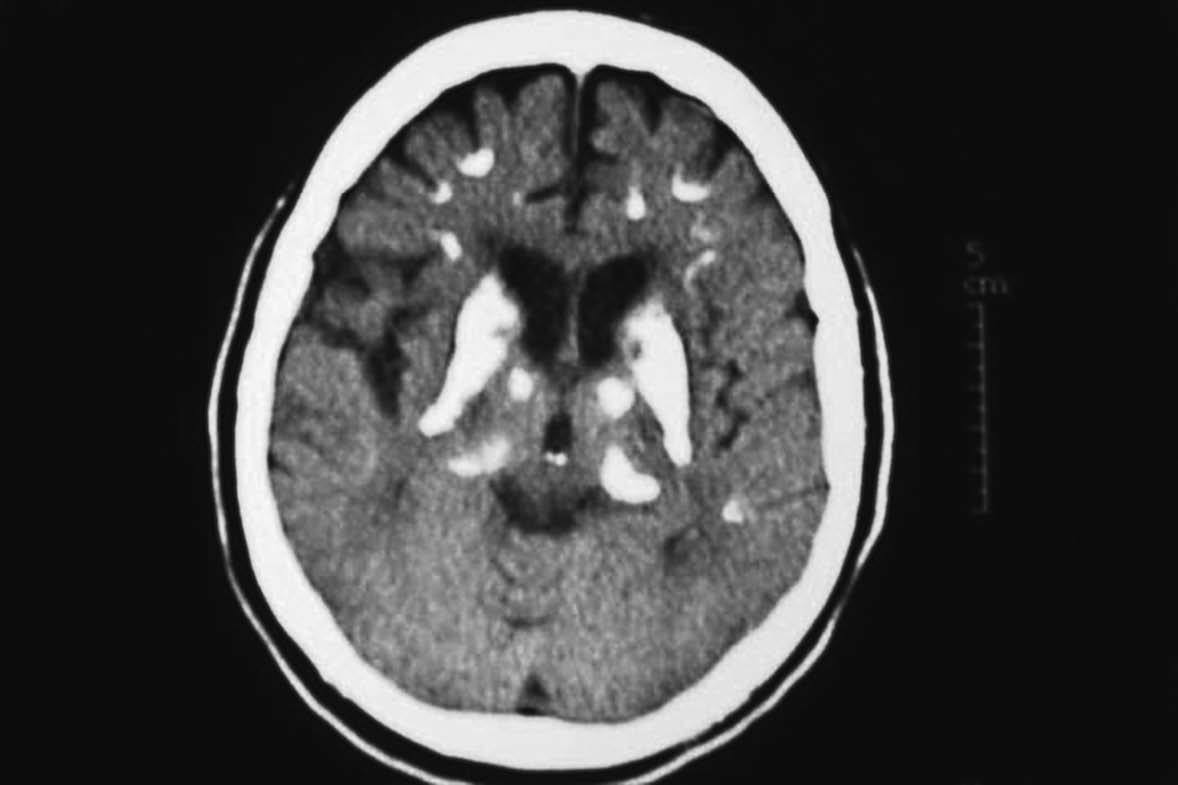
Se inició tratamiento con calcio a dosis de 1.500 mg/día y calcitriol a dosis de 0,5 µg/día, con buen control metabólico y evolución clínica satisfactoria. A los 23 años comenzó con un síndrome parkinsoniano que se trató con levodopa, con buena respuesta inicial. El paciente presentaba además una hipoacusia bilateral y simétrica neurosensorial y una hipoplasia renal izquierda. En un ingreso reciente se realizó una tomografía computarizada (TC) cerebral en la que se observaron calcificaciones bilaterales de los ganglios de la base, sustancia blanca cortical adyacente y cerebelo (fig. 1).
Fig. 1. Extensas calcificaciones cerebrales bilaterales, más prominentes en los núcleos de la base.
A pesar de un buen control metabólico, persistió el deterioro progresivo de funciones superiores y motoras así como un aumento en el tamaño y el número de las calcificaciones cerebrales. El padre, una hermana y una sobrina del paciente (la primera hija de la hermana afectada) también padecen sordera e hipoparatiroidismo primario.
Nos encontramos ante un caso de hipoparatiroidismo primario idiopático familiar con un patrón de herencia autosómico dominante con calcificaciones de los ganglios de la base bilaterales y síndrome parkinsoniano secundario, sordera neurosensorial e hipoplasia renal.
DISCUSIÓN
La primera descripción de calcificaciones en los ganglios de la base se realizó probablemente por Fahr en 19301. En 1939, se estableció la asociación entre la presencia de calcificaciones cerebrales y el hipoparatiroidismo crónico2. Sin embargo, se estima que, incidentalmente, pueden encontrarse calcificaciones en los ganglios basales en un 0,7% de las TC cerebrales. En la mayoría de los casos, sobre todo en pacientes mayores de 60 años, no suelen tener significado patológico3.
La patogenia de las calcificaciones cerebrales sigue siendo una incógnita3-5. Su estudio ha demostrado una composición química de los depósitos similar a las calcificaciones fisiológicas de hueso y dientes, tanto en las formas que se asocian a alteraciones del metabolismo del calcio y fósforo como en las de otras causas. En las enfermedades del metabolismo de calcio/fósforo, podría asociarse a anomalías en las concentraciones de estos iones a nivel intra y extracelular, si bien debe existir algún otro factor desconocido hasta el momento, ya que se han descrito casos de aumento en el tamaño de las calcificaciones en estados de normocalcemia 5.
En la mayoría de los casos, las calcificaciones de los ganglios basales son esporádicas, aunque existen formas hereditarias y familiares. Tanto las formas esporádicas como las familiares suelen estar asociadas a alteraciones del metabolismo de calcio/fósforo o de la PTH4.
El caso que presentamos asocia un hipoparatiroidismo primario con un patrón de herencia autosómico dominante, con sordera neurosensorial y displasia renal. El hipoparatiroidismo primario familiar aislado se puede heredar con un carácter autosómico dominante, recesivo o ligado al cromosoma X, aunque las calcificaciones de los ganglios basales se han visto fundamentalmente en las formas autosómicas dominantes4. En una revisión clásica, se recogen 42 casos de hipoparatiroidismo idiopático familiar, agrupados en 9 familias3. La clínica en estos pacientes se iniciaba entre los 30 y los 50 años, con dificultades para el habla, deterioro cognitivo y signos extrapiramidales, y las crisis comiciales eran raras. En este grupo de pacientes, la presencia de calcificaciones cerebrales se asociaba a síntomas neurológicos, pero ninguno de estos enfermos padecía anomalías renales o sordera.
La combinación de hipoparatiroidismo primario, sordera neurosensorial y displasia renal fue descrita por primera vez en 19926, y fue propuesto como un síndrome nuevo en 1997 con el acrónimo de HDR7. Lichtner et al8 describen 2 casos de síndrome HDR y clínica de síndrome de DiGeorge (anomalías faciales, alteraciones cardíacas, aplasia/hipoplasia paratiroidea y tímica y alteración del desarrollo psicomotor), asociados a monosomía parcial del brazo corto del cromosoma 10. Se sabe que esta anomalía genética, concretamente la hemicigosis para la región DGCR2, se asocia a manifestaciones de síndrome de DiGeorge y a síndrome velocardiofacial. La presencia de la tríada de sordera, hipoparatiroidismo primario y alteraciones renales parece asociarse a la delección de uno o varios genes que se localizan en el brazo corto del cromosoma 10 en un locus distal a la región DGCR28.
Parece, por tanto, que este caso puede tratarse de un síndrome HDR, con expresividad variable, como se ha descrito en otros casos9, si bien no se han realizado estudios genéticos para determinar la presencia de la delección a nivel del cromosoma 10.
Lo abigarrado de la clínica del paciente puede deberse al retraso diagnóstico del hipoparatiroidismo, que llevó a un deterioro neurológico irreversible con un sídrome extrapiramidal incapacitante probablemente asociado con las calcificaciones de los ganglios basales.
