


Kallmann syndrome is the most common form of isolated hypogonadotropic hypogonadism with delayed puberty. The syndrome characteristically includes GnRH deficiency associated with anosmia or hyposmia due to agenesis or hypoplasia of the olfactory bulbs.1 This is a hereditary, genetically heterogeneous disease that may be transmitted as an X chromosome-linked trait or as an autosomal dominant or recessive trait. Its incidence is approximately 1/8000 in males and 1/40,000 in females.2 It is usually diagnosed at 14–16 years of age when medical advice is sought for delayed puberty.
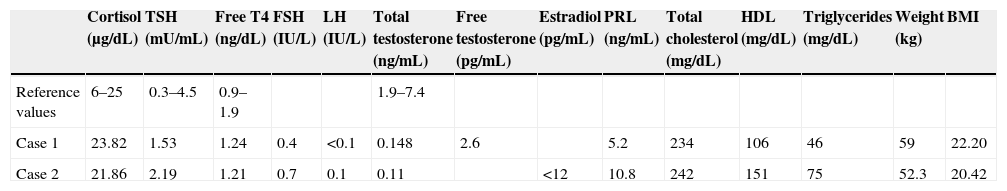
The first case reported was a 52-year-old male who was referred for severe osteoporosis with hypogonadal phenotype. His clinical history included severe hearing loss in the left ear and a cleft lip from birth. Examination revealed an absence of body hair, cryptorchidism, micropenis, and gynecomasty. The patient had previously concealed these signs. Hypogonadism was suspected based on the clinical data, and hormone and imaging tests were performed. Hormone test results (Table 1) suggested hypogonadotropic hypogonadism. Brain MRI confirmed severe hypoplasia of both olfactory bulbs. Based on the above data, the most likely diagnosis was Kallmann syndrome.
| Cortisol (μg/dL) | TSH (mU/mL) | Free T4 (ng/dL) | FSH (IU/L) | LH (IU/L) | Total testosterone (ng/mL) | Free testosterone (pg/mL) | Estradiol (pg/mL) | PRL (ng/mL) | Total cholesterol (mg/dL) | HDL (mg/dL) | Triglycerides (mg/dL) | Weight (kg) | BMI | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference values | 6–25 | 0.3–4.5 | 0.9–1.9 | 1.9–7.4 | ||||||||||
| Case 1 | 23.82 | 1.53 | 1.24 | 0.4 | <0.1 | 0.148 | 2.6 | 5.2 | 234 | 106 | 46 | 59 | 22.20 | |
| Case 2 | 21.86 | 2.19 | 1.21 | 0.7 | 0.1 | 0.11 | <12 | 10.8 | 242 | 151 | 75 | 52.3 | 20.42 |
The second case was a 44-year-old female treated with oral contraceptives since the age of 18 for primary amenorrhea, which persisted after several attempts at treatment discontinuation. In the clinical history, the patient reported anosmia. Hormone tests revealed hypogonadotropic hypogonadism, a densitometric study showed osteoporosis, and brain MRI confirmed severe hypoplasia of both olfactory bulbs.
Kallmann syndrome was first described in 1856.3 The prevalence of the syndrome is approximately four times greater in males as compared to females. From the pathophysiological viewpoint, it is explained by a defect in the migration of GnRH-secreting fetal neurons from the olfactory placode (where they originate) to the medial basal hypothalamus, where they represent the GnRH pulse generator. This defect may be absolute or partial. Because of this, cells that contain GnRH and neurites end up in a tangle around the lamina cribrosa and in the dural layers adjacent to the meninges, below the prosencephalon.7
There are several genes involved in the development of the syndrome. The best known is the KAL1 gene, located in locus Xp 22.3, an X chromosome-linked gene4 that escapes X inactivation, which is responsible for so-called Kallmann syndrome 1 (KAL1). The KAL1 gene encodes for a 680-amino acid glycoprotein called anosmin-1, with characteristics of extracellular neural adhesion, which may function as an “explorer” to guide GnRH neurons to the medial basal hypothalamus. Anosmin-1 has been shown to be distributed in the olfactory placode and prosencephalon by weeks 5–6 of fetal life. KAL1 gene deletion and/or mutations cause a frameshift and premature stop codons that result in the changes defining Kallmann syndrome.5 This change rarely has an impact on females.
Other hereditary forms are less well known. The autosomal dominant form is known as Kallmann syndrome 2 (KAL2), and the autosomal recessive form is called Kallmann syndrome 3 (KAL3).
The involvement of several genes4 may account for phenotype variability in each case. However, intrafamilial heterogeneity is also seen.6
The cases reported here showed the main two clinical characteristics, hypogonadism and anosmia.1 A detailed clinical history with a special focus on anosmia suggested the diagnosis. It should not be forgotten, however, that there may be other associated characteristics such as gynecomastia, cryptorchidism, micropenis, cleft lip, cleft palate, imperfect facial fusion, seizures, short metacarpal bones, pes cavus, sensorineural hearing loss, cerebellar ataxia, etc., some of which were found in the first patient.
In patients with hyposmia, smell tests are often difficult to interpret. Coronal and axial cranial MRI of olfactory sulci-bulbs provides greater sensitivity, showing olfactory bulb aplasia or hypoplasia in approximately 90% of cases and leading to diagnosis, especially in small and prepubertal children. With MRI, olfactory sulci may be detected from week 30 of pregnancy, and olfactory bulbs between weeks 30 and 34.8 Genetic study is very important, and could almost be considered as mandatory, but was not performed in the reported cases due to patient refusal.
According to the literature, most cases are usually diagnosed at 14–16 years of age.8,9 The uniqueness of our cases lies in their being diagnosed at such a late age. In the first case, late diagnosis was due to a psychosocial and cultural component which had previously prevented the patient from consulting a specialist. By contrast, our second patient consulted a doctor at 18 years of age, but the etiology of primary amenorrhea was not investigated at the time, and was simply treated as a symptom.
When hypogonadism is diagnosed late, its manifestations include absent or poor sexual development inappropriate for the patient's age, eunuchoid proportions due to the late closure of growth cartilages in limbs with a reduced relationship between the upper and lower segment and increased arm span, osteopenia/osteoporosis due to decreased bone matrix mineralization and increased bone resorption, and body composition changes with increased fat mass (including abdominal mass) of gynoid distribution and decreased muscle mass. Late treatment of hypogonadism thus leads to an increase in fracture and cardiovascular risks, and also to earlier aging.
It may be concluded that Kallmann syndrome is an uncommon condition that should be suspected in patients with hypogonadotropic hypogonadism irrespective of age, and that special attention should be paid in the clinical history to the presence of hyposmia.1 This will allow for an adequate diagnosis to be made and, consequently, for early replacement therapy to be started, so decreasing all potential consequences of the deficiency.
Please cite this article as: Cabrejas Gómez MC, Vicente Vicente MÁ, Antón Miguel MÁ, Urcelay Rojo M. Síndrome de Kallmann de diagnóstico tardío. Endocrinol Nutr. 2015;62:106-108.
