


El síndrome de Kallmann es la forma más frecuente de hipogonadismo hipogonadotropo aislado con pubertad retrasada. Característicamente existe déficit de GnRH asociado a anosmia o hiposmia por agenesia o hipoplasia de los bulbos olfatorios1. Se trata de una enfermedad hereditaria, genéticamente heterogénea, pudiéndose transmitir como rasgo ligado al cromosoma X o de forma autosómica dominante o recesiva. Su incidencia aproximada es de 1/8.000 en varones, y 1/40.000 en mujeres2. Su diagnóstico suele ser entre los 14-16 años de edad, cuando se consulta por retraso puberal.
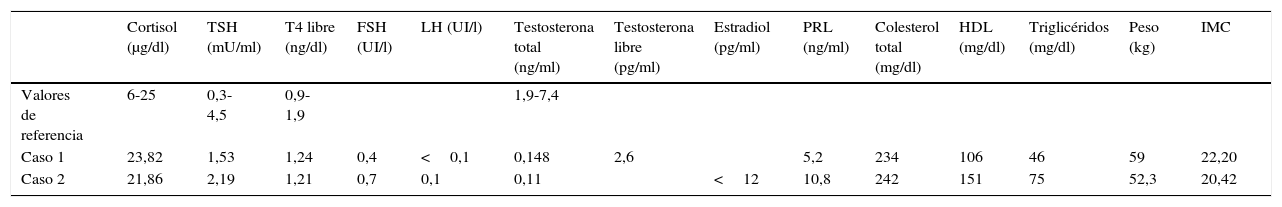
El primer caso se trata de un varón de 52 años, que fue remitido por presentar osteoporosis grave con fenotipo hipogonadal. Como antecedentes destacaban una hipoacusia severa izquierda y un labio leporino desde el nacimiento. A la exploración se objetivaron ausencia de vello junto con criptorquidia, micropene y ginecomastia. El paciente lo había ocultado hasta entonces. Ante los datos clínicos presentes se sospechó hipogonadismo y se llevaron a cabo estudios hormonales y de imagen. Los resultados del análisis hormonal (tabla 1) fueron de hipogonadismo hipogonadotropo. En la RMN cerebral se confirmó una hipoplasia severa de ambos bulbos olfatorios. Ante los datos expresados el diagnóstico más probable es el de síndrome de Kallmann.
| Cortisol (μg/dl) | TSH (mU/ml) | T4 libre (ng/dl) | FSH (UI/l) | LH (UI/l) | Testosterona total (ng/ml) | Testosterona libre (pg/ml) | Estradiol (pg/ml) | PRL (ng/ml) | Colesterol total (mg/dl) | HDL (mg/dl) | Triglicéridos (mg/dl) | Peso (kg) | IMC | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Valores de referencia | 6-25 | 0,3-4,5 | 0,9-1,9 | 1,9-7,4 | ||||||||||
| Caso 1 | 23,82 | 1,53 | 1,24 | 0,4 | <0,1 | 0,148 | 2,6 | 5,2 | 234 | 106 | 46 | 59 | 22,20 | |
| Caso 2 | 21,86 | 2,19 | 1,21 | 0,7 | 0,1 | 0,11 | <12 | 10,8 | 242 | 151 | 75 | 52,3 | 20,42 |
Por otro lado, en un segundo caso estudiamos una mujer de 44 años, tratada desde los 18 años con anticonceptivos orales por amenorrea primaria, con persistencia de la misma tras varios intentos de retirada del tratamiento. En la anamnesis refería anosmia. Tras realizarle análisis hormonal se objetivó un hipogonadismo hipogonadotropo, en el estudio densitométrico se observó una osteoporosis y en la RMN cerebral se confirmó una hipoplasia de bulbos olfatorios.
El síndrome de Kallmann fue descrito por primera vez en 18563. La prevalencia es aproximadamente 4 veces mayor en los varones que en las mujeres. Desde el punto de vista fisiopatológico, se explica por un defecto en la migración de las neuronas fetales secretoras de GnRH desde la placoda olfatoria (donde se originan) hasta el hipotálamo medial basal, donde constituyen el generador de pulsos de GnRH. Este defecto puede ser absoluto o parcial. Debido a ello las células que contiene GnRH y las neuritas terminan en una maraña alrededor de la lámina cribosa y en las capas durales adyacentes a las meninges, por debajo del prosencéfalo7.
Hay varios genes implicados en su desarrollo. El mejor conocido es el gen KAL1 localizado en el locus Xp 22.3, gen ligado al cromosoma X4 que escapa a la inactivación X, que sería responsable del llamado síndrome de Kallmann 1 (KAL1). El gen KAL1 codifica una glucoproteína de 680 aminoácidos, llamada anosmina-1, con características de molécula de adherencia neural extracelular que podría funcionar como un «explorador» para guiar las neuronas GnRH hasta el hipotálamo medial basal. Se ha demostrado que la anosmina-1 se distribuye por la placoda olfatoria y el prosencéfalo en la semana 5-6 de vida fetal. La delección y/o mutaciones del gen KAL1 producen un cambio de marco y codones de parada prematura que daría lugar a las alteraciones que definen el síndrome de Kallmann5. Esta alteración rara vez repercute en las mujeres.
Otras formas hereditarias son menos conocidas. La forma autosómica dominante se conoce como síndrome de Kallmann 2 (KAL2), y la de herencia autosómica recesiva se conoce como síndrome de Kallmann 3 (KAL3).
El hecho de que haya varios genes implicados4 podría justificar la variabilidad del fenotipo en cada caso; sin embargo, también se observa una heterogenicidad intrafamiliar6.
En los casos aquí comentados existían las 2 características clínicas principales, hipogonadismo y anosmia1. Una historia clínica detallada incidiendo en el aspecto de la olfacción nos puso en la pista de su diagnóstico. Pero además, no hay que olvidar que pueden existir otras características acompañantes como ginecomastia, criptorquidia, micropene, labio leporino, paladar hendido, fusión facial imperfecta, trastornos convulsivos, metacarpianos cortos, pie cavo, sordera neurosensorial, ataxia cerebelosa, etc., algunas de las cuales estaban presentes en el primer caso.
Las pruebas de olfacción, en los casos de hiposmia, a veces son difíciles de interpretar, por lo que aporta una mayor sensibilidad la RMN coronal y axial craneal de los surcos-bulbos olfatorios. En ella queda patente una aplasia o hipoplasia del bulbo olfatorio en alrededor del 90% de casos, llevando al diagnóstico, especialmente en niños pequeños y de edad prepuberal. Con la RMN, los surcos olfatorios pueden detectarse desde la semana 30 de edad gestacional y los bulbos olfatorios entre la semana 30 y 348. El estudio genético es muy importante, y casi se podría decir obligado, pero en los casos mencionados no se realizó por la negativa de los pacientes.
La mayoría de los casos, según datos bibliográficos, se suelen diagnosticar a los 14-16 años8,9. La particularidad de nuestros casos es la edad tardía del diagnóstico. En el primer caso, el diagnóstico tardío fue debido a un componente psico-socio-cultural, por el cual, el paciente no consultó antes con un especialista. En cambio, en el segundo caso la paciente sí consultó a los 18 años, pero no se buscó en ese momento la etiología de la amenorrea primaria, y simplemente fue tratado el síntoma.
Cuando el diagnóstico es tardío, el hipogonadismo se manifiesta por una falta o escasez del desarrollo sexual inapropiado para la edad, proporciones enucoides por cierre tardío de los cartílagos de crecimiento en extremidades con una relación entre el segmento superior/segmento inferior reducida y una envergadura aumentada, osteopenia/osteoporosis por disminución de la mineralización de la matriz ósea y aumento de la resorción ósea, y alteraciones en la composición corporal con aumento de masa grasa (incluida abdominal) de distribución ginoide y disminución de masa muscular. De ese modo, el tratamiento tardío del hipogonadismo conduciría a un aumento del riesgo de fractura y cardiovascular, así como a un envejecimiento más precoz.
Podemos concluir que el síndrome de Kallmann, a pesar de ser una enfermedad poco frecuente, debe sospecharse en pacientes que presentan hipogonadismo hipogonadotropo independientemente de la edad, e incidir en la anamnesis preguntando sobre la presencia de hiposmia1. De esa manera se podrá llegar a un diagnóstico adecuado y, por tanto, establecer el tratamiento sustitutivo precoz, que disminuirá todas las posibles consecuencias de dicho déficit.
