



Acromegaly is a disease diagnosed with increasing frequency1 and whose mean age of onset coincides to a great extent with the child-bearing life of women.2 Treatments currently available for acromegaly include surgery, drug treatment and, in some cases, radiotherapy. These therapeutic modalities achieve tumor growth control and minimize the clinical consequences of hypersomatotropism in most patients.3
Despite the positive impact of current treatments on survival and quality of life, pregnancy in acromegaly continues to be an uncommon event and a challenge for clinicians. The largest case series reported required multicenter collaboration in France and included 59 pregnancies4, while hardly one hundred pregnancies have been reported in other publications.4 The lack of retrospective data makes it difficult to anticipate potential complications. Such information is required to provide informed counsel to patients with acromegaly who want to bear children. We therefore report a patient with active acromegaly after surgery attending our department who completed a full-term pregnancy.
A 31-year-old woman attended the gynecology clinic in 2005 reporting oligomenorrhea and galactorrhea for approximately one year, with no hirsutism or other symptoms. Available biochemical test results included prolactin levels of 86.22ng/mL (4.8–23.3ng/mL) in the absence of drugs that could affect such measurement. Pituitary magnetic resonance imaging (MRI) revealed a pituitary macroadenoma with suprasellar extension and invasion of the right cavernous sinus. The patient was referred to the neurosurgery department for surgery, which was performed in 2006 by a transsphenoidal approach with no complications. She was subsequently evaluated at our department for postoperative follow-up. The patient had not shown at any time acral thickening, prognathism, diaphoresis, headache, arthralgia, high blood pressure, hyperglycemia, or other evidence suggesting acromegaly. However, basal hypothalamic-pituitary tests (Table 1) found consistently elevated IGF-I levels and there was an absence of growth hormone (GH) suppression in an oral glucose tolerance test. The pituitary profile also included decreased serum cortisol levels, for which the patient was treated with oral hydrocortisone. Additional tests performed included an echocardiogram, a polysomnographic study, and a colonoscopy, none of which suggested changes associated with acromegaly. Postoperative MRI revealed a residual tumor in the sella turcica and right cavernous sinus, and repeat surgery was decided upon after consultation with the neurosurgery department.
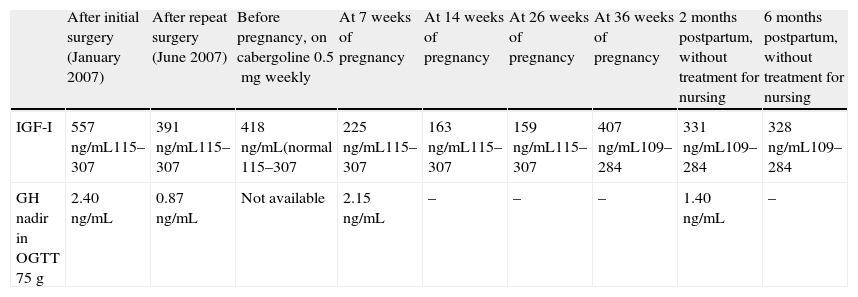
Laboratory test results of the patient.
| After initial surgery (January 2007) | After repeat surgery (June 2007) | Before pregnancy, on cabergoline 0.5mg weekly | At 7 weeks of pregnancy | At 14 weeks of pregnancy | At 26 weeks of pregnancy | At 36 weeks of pregnancy | 2 months postpartum, without treatment for nursing | 6 months postpartum, without treatment for nursing | |
| IGF-I | 557ng/mL115–307 | 391ng/mL115–307 | 418ng/mL(normal 115–307 | 225ng/mL115–307 | 163ng/mL115–307 | 159ng/mL115–307 | 407ng/mL109–284 | 331ng/mL109–284 | 328ng/mL109–284 |
| GH nadir in OGTT 75g | 2.40ng/mL | 0.87ng/mL | Not available | 2.15ng/mL | – | – | – | 1.40ng/mL | – |
GH: growth hormone; IGF-I: insulin-like growth factor-I; OGTT: oral glucose tolerance test.
Endoscopic transsphenoidal surgery was performed at our hospital, and a significant decrease in IGF-I levels and tumor mass reduction were achieved. The pathological laboratory reported an adenoma with immunohistochemistry positive for GH and negative for prolactin, with a Ki 67 index of 5%. After surgery, the patient achieved regular menses and had no evidence of hypopituitarism. However (Table 1), biochemical criteria of disease activity persisted, as well as gross tumor remnants. Because of a slight increase in IGF-I levels, treatment was attempted with cabergoline, which improved biochemical control of the disease. At treatment start, the patent had been recommended to avoid pregnancy until biochemical normalization and tumor stability had been ensured.
In 2008, three months after the final control and at 35 years of age, the patient attended our department reporting a five-week pregnancy that she wanted to continue. Therefore, cabergoline treatment was discontinued, and three-monthly neurological and ophthalmological controls and monitoring of high-risk pregnancy at the clinic were scheduled. The patient was also advised to stop smoking, which she declined to do. She remained symptom-free during pregnancy until she delivered a healthy baby at 39 weeks. At the controls made (Table 1), IGF-I levels decreased during pregnancy to levels normal for age, except in the last measurement. The O'Sullivan test was normal in the first and third trimesters, and no treatment was required for blood pressure. At the end of pregnancy, MRI showed tumor stability (Fig. 1). However, postpartum IGF-I levels again exceeded physiological limits, although the patient reported no symptoms. In order to decrease the morbidity associated with excess GH, the patient started treatment with cabergoline, which led to a gradual improvement in the biochemical parameters and to tumor stability.
 and after (B) pregnancy.")
Acromegaly is a syndrome with high associated morbidity and mortality. Fortunately, we now have available an increasingly wide therapeutic armamentarium to improve patient prognosis and quality of life. However, despite these advances, little experience is available in pregnancy monitoring in patients with acromegaly. There may be several reasons for this:
- -
Hyperactivity of the somatotropic axis may alter hormonal cycles leading to ovulation. This, combined with the greater frequency of ovarian functional hyperandrogenism and increased insulin resistance in patients with acromegaly,2,3,5 may hinder ovulation and, thus, pregnancy. In addition, hypogonadotropic hypogonadism due to tumor compression or iatrogenic in nature (surgery and radiotherapy) may coexist, and will then require external hormonal induction of ovulation to achieve pregnancy.
- -
The burden of disease symptoms and the limited experience with most drugs available for acromegaly in pregnancy may discourage attempts at pregnancy in patients and their physicians. Discontinuation of somatostatin analogues and cabergoline is currently recommended when pregnancy is documented, but the clinical cases reported have not shown that these drugs are teratogenic or increase maternal or fetal risk. In addition, surgery involves a risk of pregnancy interruption, and radiotherapy is directly contraindicated in pregnant patients.
- -
The greater difficulty in the clinical and biochemical monitoring of the disease: during pregnancy, pituitary GH (GH1) secretion is physiologically replaced by placental GH (GH-V) secretion; healthy patients suppress GH1 secretion, while pregnant women with acromegaly maintain steady GH1 levels. In addition, gestational hypoestrogenism blocks IGF-I secretion in the liver, which explains the decreased IGF-I secretion in our patient.5,6 Moreover, the different GH variants and IGF-I do not cross the placental barrier, which prevents their having direct harmful effects on the fetus.7 The abovementioned physiological changes do not allow for the reliable monitoring of acromegaly in pregnancy based on these biochemical parameters, and the physical changes inherent in pregnancy (edema, limb swelling, etc.) may be confounded with disease symptoms. In addition, MRI should be performed without contrast, and only in cases with symptoms of potential visual involvement, which increases the uncertainty regarding the clinical course.
Based on the above considerations, the following should be taken into account when a patient with acromegaly inquires about the feasibility of a future pregnancy:
- 1)
The patient should be monitored for tumor growth during pregnancy, and the specific risks to pregnancy of the available treatments (surgery, somatostatin analogues, and cabergoline) should be taken into account.
- 2)
The patient should be monitored closely for gestational diabetes and pregnancy-induced hypertension, each with its own specific maternal and fetal risks, particularly in the presence of active disease.
- 3)
There is no evidence that excess growth hormone and IGF-I on their own increase the maternal and fetal risks.
- 4)
Biochemical monitoring is of less value during pregnancy, and clinical data are more difficult to interpret, because the typical changes of pregnancy may be very similar.
Despite the above, the experiences reported to date have been favorable in a majority of cases, although a publication bias cannot be ruled out. In the largest series reported to date,4 including 59 pregnancies, most patients were able to safely discontinue drug treatment. The offspring of these patients included six microsomic and two macrosomic fetuses, and there was a slightly higher incidence of gestational diabetes and arterial hypertension induced by pregnancy, particularly in patients with active disease. Tumor volume remained stable in most patients. Despite these positive data, the currently available experience regarding pregnancy in acromegaly is scant, and consistent and comprehensive analysis of data from future pregnancies is therefore required, preferably in the setting of multicenter studies. Such studies would allow for more rigorous counseling about the feasibility of pregnancy in patients with acromegaly, which should be part of the treatment of childbearing patients with any pathological condition.
Please cite this article as: Fernández A, et al. Seguimiento de gestación en paciente con acromegalia: descripción de caso clínico y revisión de la literatura. Endocrinol Nutr. 2013;60:102–5.
